

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pyruvate dehydrogenase kinase (PDK) participates in the regulation of the pyruvate dehydrogenase (PDH) complex (PDC), in which PDH is the first component. PDK is a kinase enzyme that acts to inactivate the enzyme PDH by phosphorylating it using adenosine triphosphate (ATP). Four isomeric forms of PDK, namely PDK1-4, exist having tissue specific expression, different activities, and dissimilar phosphorylation rates (see [1] for comprehensive review). PDC, which acts as one of the major enzymes responsible for the regulation of glucose metabolism, is a nuclear-encoded mitochondrial multienzyme complex that catalyzes oxidative decarboxylation of pyruvate to form acetyl-coenzyme A (CoA) and thereby provides the primary link between glycolysis and the tricarboxylic acid (TCA) cycle [2]. A phosphorylation/dephosphorylation cycle regulates the enzymatic activity of PDH, and phosphorylation results in the inactivation of PDH. PDC is composed of three catalytic components namely, PDH (E1), dihydrolipoamide transacetylase (E2) and dihydrolipoamide dehydrogenase (E3). These components are organized into large multimeric complexes together with the structural subunit E3 binding protein. The basic core of the E1 PDH component is a heterotetramer of two alpha and two beta subunits (α2β2), and it catalyzes the first step of pyruvate decarboxylation. PDKs inhibit PDC by catalyzing the phosphorylation of serine residues in the E1 alpha subunit. PDH is one of the most important and pivotal dehydrogenases having control over mitochondrial metabolic pathways and catalyzes the irreversible decarboxylation of pyruvate to acetyl-CoA, CO2 and nicotinamide adenine dinucleotide (reduced form) [NAD(H)]. Because PDH controls the entry of carbon into the TCA cycle, regulation of PDH activity governs the entry of carbons derived from carbohydrates into the mitochondria. The reaction has important roles not only in the regulation of mitochondrial energy-producing pathways [TCA and oxidative phosphorylation (OXPHOS)], but also in the generation of biosynthetic intermediates, such as citrate. Glucose is the major source of carbon for mammalian cells, including cancerous cells like gliomas. Glucose is metabolized to generate ATP, through cytosolic glycolysis and oxygen-dependent mitochondrial metabolism, in which most of the reducing potential is the outcome of the TCA cycle. The entry of glucose into the TCA cycle is controlled by PDH. Thus, the proper functioning of mitochondrial metabolic pathways can only be achieved through the continuous real-time control of PDH. PDK downregulates the activity of PDH and decreases the oxidation of pyruvate in mitochondria and increases the conversion of pyruvate to lactate in the cytosol. Because these regulatory processes in numerous pathological conditions are extensively altered, these alterations may reflect targets for therapeutic interference.
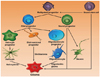
Malignant gliomas are the most frequent primary brain tumors that arise from the supportive non-neuronal cells of the brain, called glial cells [3-5]. Malignant gliomas contain multipotent tumor stem cells having potential to be transformed into variants of normal neural progenitor cells that are responsible for populating and repopulating the tumors [6,7]. Around 30 percent of all brain and central nervous system tumors and 80 percent of all malignant brain tumors are gliomas [8]. In contrast to neurons, glial cells have the potency to divide and multiply, and failure due to any reason in the controlling system of this potency results in the formation of a glioma. Malignant gliomas are among the most fatal human cancers [9,10]. Gliomas are characterized by their infiltrating nature, especially into the surrounding normal brain tissue. Similar to other human cancers, the formation and progression of diffuse gliomas is accompanied by the overexpression of growth factors like platelet-derived growth factor, epidermal growth factor receptor, basic fibroblast growth factor, transforming growth factor-alpha, and insulin-like growth factor-1 causing an autocrine growth-promoting loop, loss of cell cycle control, activation of oncogenes, inactivation of tumor suppressor genes, dysregulation of apoptosis and instability of the genome. The histological presence of microvascular proliferation is also an important identifying feature of high grade malignant gliomas. Malignant gliomas, primarily glioblastomas, contain angiogenic molecules like vascular endothelial growth factor (VEGF) suggesting an "angiogenic switch" for the progression to malignant gliomas [4,11]. Physiologic response to hypoxia causes the occurrence of tumor angiogenesis through the increased transcription of the VEGF gene by the hypoxia-inducible factor (HIF) family of transcription factors [12-14]. Their highly infiltrative nature is one of the major challenges that prevents surgical resections and complicates the effective delivery of several therapies. The augmentation of the tumor in histological grade creates additional features of malignancy. Gliomas are named on the basis of the cell type, with which they share histological characteristics. They are named as ependymomas, astrocytomas (glioblastoma multiforme), oligodendrogliomas and mixed gliomas (e.g., oligoastrocytomas) on the basis of their resemblance with ependymal cells, astrocytes, oligodendrocytes and mixed glial cells, respectively (Fig. 1). Malignant gliomas include glioblastomas [World Health Organization (WHO) grade IV], anaplastic astrocytomas (WHO grade III), mixed anaplastic oligoastrocytomas (WHO grade III), and anaplastic oligodendrogliomas (WHO grade III).
DYSREGULATED METABOLISM AS A HALLMARK OF GLIOMAGENESIS
Growing evidence reveals that all cancers regardless of tissue or cellular origin are a disease of impaired cellular energy metabolism [15]. In addition to the previously well recognized hallmarks of cancers [16-19], aerobic glycolysis or the Warburg effect is also a robust metabolic feature of most tumors [20-24]. Recent studies on gliomas in experimental models show the dependence of glioma cells on glycolysis as the primary source of energy [25]. Upregulated glycolysis has been established as a defining feature of primary and metastatic cancers that results in increased glucose consumption [23]. Malignant gliomas display high rates of glycolysis and lactate production, even in the presence of adequate oxygen, a phenomenon well known as aerobic glycolysis or the Warburg effect. On the other hand, tumor hypoxia results in constitutive upregulation of glycolysis and acidosis, contributing to the tumor resistance to therapeutic agents [23,26]. The progression of gliomagenesis often occurs in a hypoxic microenvironment that compels the use of anaerobic glycolysis as the primary energy source [23]. Hypoxia stabilizes HIF, a transcription factor, which increases the biological aggressiveness of tumors, promoting glycolysis, cellular proliferation, and angiogenesis [27-30]. Once activated, HIF can regulate the expression of many glycolytic enzymes including glucose transporters and mitochondrial enzymes that are involved in the metabolic adaptation to hypoxia through the conversion of glucose to pyruvate and subsequently to lactate [31]. Abnormal energy metabolism in gliomas is also linked to apoptotic signaling in the gliomas. Suppressing mitochondrial activity may give cancer cells a proliferative advantage by suppressing apoptosis. Understanding the defects in mitochondrial energy metabolism and intrinsic apoptotic signaling pathways in gliomas can provide novel therapeutic strategies [32].
ROLE OF PDKS IN GLIOMAS
Regulation of glucose metabolism by PDK in gliomas: altered energy metabolism
Tumor cells show an increased dependence on glycolysis for ATP production, even in the presence of plentiful oxygen [33,34]. More than half a century ago, Otto Warburg postulated that cancer cells predominantly produce ATP through a high rate of glycolysis followed by lactic acid fermentation in the cytosol, rather than by the comparative low rate of glycolysis followed by oxidation of pyruvate in mitochondria in non-cancerous cells [23,35]. This change in metabolism was postulated as a fundamental cause of cancer (Warburg effect) [36,37]. Griguer et al. [38] have investigated the specific bioenergetic markers associated with the metabolic phenotypes of numerous glioma cell lines. They speculated that heterogeneity existed in glucose metabolism and identified important biological markers in glioma cells that are critical for the progression of gliomas. Similarly, Morfouace et al. [39] have recently investigated the difference in glucose metabolism between rat glioma stem cells and neural stem cells. Mitochondrial inactivation in cancer is predominantly due to the inhibition of PDH by PDK. NAD+ required for a high glycolytic flux is generated by the conversion of pyruvate into lactate. Recently van Horssen et al. [40] have reported based on the genetic and pharmacological inhibition of nicotinamide phosphoribosyltransferase in glioma cells that the fluctuation in intracellular [NAD(H)], which occurs in response to the reprogramming of cellular metabolism, significantly affects cell growth and invasion, suggesting that nutrient- or drug-mediated modulation of NAD(H) could be a new possible option for controlling the motility and invasion of glioma cells. The upregulated level of lactate is considered as a cause for a poor prognosis in several cancers including malignant gliomas [41,42]. This has resulted in an extensive search to identify glycolysis inhibitors for use in cancer therapy. Dichloroacetate (DCA), a PDK inhibitor, has fuelled prospects for such metabolic targeting, and further investigations are ongoing to validate the potential of metabolic targeting as a cancer therapy [43].
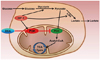
Alterations in cellular energy metabolism, the glycolytic pathway in particular, are one of the important distinguishable features of cancer cells. Pyruvate is a critical metabolic checkpoint because it can have two fates. It can either be converted to acetyl CoA by PDH to enter into the TCA cycle or be transformed to lactate by lactate dehydrogenase (Fig. 2). PDH is the pivotal enzyme that can characterize cancer cell metabolism based on the two alternative PDH substrates available in cancer cells, namely pyruvate and acetaldehyde [44]. Glycolytic metabolism is upregulated in gliomas [45,46]. In normal cells, PDH, associated with the inner mitochondrial membrane, decarboxylates pyruvate to acetyl-CoA, which then feeds into the TCA cycle to produce citrate in the mitochondria. Consequently, PDH links the cytosolic glycolytic pathway to the mitochondrial TCA cycle. On the other hand, the activity of the PDH decreases in cancer cells and turns itself into a non-oxidative decarboxylase. The reduced activity of PDH is mainly due to the binding and phosphorylation of its subunits by a regulatory enzyme PDK [47]. PDK modulates PDH by phosphorylating three serine residues in the alpha subunit of PDH-E1 [47], which results in reduced activity. PDK activation shunts pyruvate away from the mitochondria leading to decreased flux through the TCA cycle, and subsequently the reduced release of NADH and FADH2 to the electron transport chain.
Regulation of PDK expression and mitochondrial respiration by hypoxia inducible factor (HIF) in gliomas
The uptake of glucose by metastatic cancer cells remarkably increases compared to non-cancerous cells, which causes a switch from the oxygen-dependent metabolic pathway, the TCA cycle, to an oxygen-independent pathway, glycolysis under hypoxic conditions [48]. Hypoxia in solid tumors plays an important role in the propagation of a cascade of molecular pathways in favor of tumor growth [49]. Hypoxia-induced HIFs stimulate angiogenesis and increase malignancy, metastasis, and resistance to therapy [50]. HIF-1α is one of the key factors that regulates the expression of VEGF, which plays an important role in angiogenesis in gliomas [51]. Under hypoxic condition, mammalian cells undergo a metabolic change in which glucose consumption is elevated and glycolytic pyruvate is redirected to lactate, in order to enable net ATP production by an oxygen-independent mechanism [52]. This shift from an oxidative to a glycolytic metabolism helps to maintain redox homeostasis, cell growth and survival. This metabolic shift is mediated by HIFs that control the transcription of key enzymes involved in glucose metabolism [53]. HIF-1 promotes glycolysis by upregulating the expression of glucose transporters and glycolytic enzymes. NAD+ regeneration is coupled with pyruvate reduction to lactate which enables continued glycolysis and ATP production in hypoxic cells. HIF-1 activates PDK which shunts pyruvate away from the mitochondria. HIF-1-induced PDK inhibits PDH and blocks the conversion of pyruvate to acetyl CoA resulting in decreased flux through the TCA cycle. The compromised TCA cycle activity causes the attenuation of oxidative phosphorylation and mitochondrial reactive oxygen species (ROS) production. This shut-down of the formation of mitochondrial acetyl-CoA and OXPHOS contributes to the increase in lactate, which acidifies the microenvironment leading to increased invasion and migration. HIF-1α plays a crucial role in the progression of malignant gliomas that are characterized by their effective immune escape mechanisms. Recently, Tong et al. [54] have reported that hypoxia increases cell invasion through the time-dependent expression of HIF-1α in human glioma cells.
PDK overexpression in hypoxia attenuates ROS generation and inhibits cell death due to hypoxia-induced apoptosis [55]. Here, PDK serves as a gate-keeping enzyme, which regulates pyruvate flux from the cytoplasm into the mitochondria, the site for glucose oxidation. PDK prevents the coupling of glycolysis to glucose oxidation by inhibiting its target enzyme PDH. Consequently, glycolysis is completed in the cytoplasm, where further metabolic transformation of pyruvate occurs without being oxidized in the mitochondria. This metabolic shift is a primary response in most tissues having a hypoxic adaptation. Therefore, elements involved in the hypoxia-response pathway can be a promising candidate for therapeutic targeting in gliomas.
METABOLIC MODULATION THROUGH PDK INHIBITION: A THERAPEUTIC APPROACH IN THE TREATMENT OF MALIGNANT GLIOMAS
Malignant gliomas in addition to several other solid tumors develop resistance to cell death in part through the switch from mitochondrial oxidative phosphorylation to cytoplasmic glycolysis. Identification of aerobic glycolysis as a therapeutic target offers a novel means for the treatment of gliomas. PDH is the key regulator of cellular metabolism, which is inhibited by PDK. Inhibition of the enzyme PDH leads to the conversion of pyruvate into lactate. High levels of lactate are closely associated with a poor prognosis in various types of cancers including malignant gliomas [41,56]. It necessitates the identification of glycolysis inhibitors for the treatment of malignant gliomas.
Several recent reports have placed DCA, a molecule used for over two decades in the treatment of certain pediatric mitochondrial diseases, as a PDK inhibitor to treat malignancies [57]. DCA is a pyruvate mimetic compound, which stimulates mitochondrial function by inhibiting PDKs. Inhibition of PDKs by DCA reactivates PDH and allows pyruvate to be oxidized in the mitochondria. Simultaneously, this induces apoptosis in cancer cells, with little or no effect on non-cancerous cells. DCA increases apoptosis in malignant glioma cells. The crystal structure of PDK2 in complex with DCA shows that DCA occupies the pyruvate binding site in the N-terminal regulatory (R) domain of PDK [58]. It has been reported that PDK2 is the most sensitive and PDK3 the most resistant, while PDK1 and PDK4 are relatively sensitive to DCA inhibition [59,60]. Wong et al. [61] have reported that DCA is an effective sensitizer in most endometrial cancer cell lines to apoptosis through nuclear factor of activated T-cells, Kv1.5 and p53 upregulated modulator of apoptosis-mediated mechanisms. DCA can promote a metabolic shift from glycolysis to oxidative phosphorylation [62], thereby inducing apoptosis by lowering the mitochondrial membrane potential, activating potassium channels and inhibiting tumor cell growth [57,63,64]. Although there are some reports describing controversies for DCA in its antitumor efficacy versus toxicity in normal tissues in vitro and in vivo, evidence supports the effective role of DCA in killing several types of cancer cells [43,57,65-69]. Michelakis et al. [70] have recently tested the potency of DCA to reverse cancer-specific metabolic and mitochondrial remodeling in glioblastoma, suggesting metabolic modulation through PDK inhibition as a novel therapeutic strategy for the treatment of glioblastoma multiforme (GBM). DCA treatment rapidly reversed mitochondrial hyperpolarization in GBM. DCA therapy induced apoptosis and increased mitochondrial reactive oxygen species in GBM by inhibiting HIF-1α, promoting p53 activation and suppressing angiogenesis. Up until now, gliomas have been one of the most dreadful malignant tumors with no successful treatment [71]; this discovery opens new therapeutic possibilities. Morfouace et al. [39] have recently studied the impact of DCA on the apoptotic pathway in a rat model. The authors have suggested the dependence of early tumorigenesis on glycolysis, and they have provided the rationale for the combination of DCA with conventional treatments to eradicate cancer stem cells and to prevent tumor relapse while preserving neural stem cells. Recently, Duan et al. [72] have investigated the antitumor activity of DCA in C6 rat glioma cells in vitro and in vivo. They found that DCA inhibited C6 glioma cell proliferation, induced C6 cell apoptosis, arrested cell cycle in S phase, induced ROS production, decreased the mitochondrial membrane potential in tumor tissues and finally exhibited anti-angiogenic effects. These findings suggest that PDK could be a promising therapeutic target for the treatment of malignant gliomas. Recent reports have suggested phenylbutyrate as a possible and a safer alternative to DCA as a PDK inhibitor [73,74].
CONCLUDING REMARKS
Multiple layers of metabolic alterations play an important role in the proliferation and survival of cancer cells. Evidence documented in recent years indicates the reprogrammed glioma cell metabolism could be exploited as a therapeutic target of malignancies. HIF-induced PDK plays a pivotal role in regulating mitochondrial activity. PDK acts as a gate-keeping mitochondrial enzyme that downregulates PDH activity, decreases the oxidation of pyruvate in mitochondria, and increases the conversion of pyruvate to lactate in the cytosol. Increased tumorigenicity correlates with higher PDK activity, lower PDH activity and a dependence on glycolytic pathways. Thus, the metabolic targeting of PDK can be a fruitful effort in designing treatment strategies that can slow down glioma progression and improve the response to therapy. PDK inhibition could result in a positive clinical outcome in the treatment of malignant gliomas with greater sensitivity to glycolytic inhibitors. Investigations on the impact of specific PDK inhibitors (for example, DCA and phenylbutyrate) on the metabolic network as a whole, including fatty acids, nucleotides and amino acid metabolism, needs to be conducted in the future for toxicity-free and targeted treatment of malignant gliomas.
 XML Download
XML Download