

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Central nervous system (CNS) germ cell tumors (GCTs) represent about 3% of primary pediatric brain tumors in Western countries, but up to 11% in Asian countries [1,2]. CNS GCTs encompass a wide pathologic spectrum: the majority are germinomas (-55%) and teratomas and mixed GCTs (-33%), and the remaining 10% are malignant endodermal sinus tumors, embryonal cell carcinomas, choriocarcinomas, and teratocarcinomas. The most common location is the pineal region, followed by the suprasellar regions of the brain [1,2].
With the exception of small-to-moderate intratumoral cysts, germinomas appear on magnetic resonance imaging (MRI) as a well-demarcated mass with relatively uniform signal intensity [3]. Mixed GCTs and teratomas demonstrate an extensive mixture of elements, may have both hyperintense and hypointense regions on T2-weighted images, and may show heterogeneous enhancement [4].
In a study that analyzed the MRI features of GCTs, intracranical GCTs with a cystic component were not uncommon [5]. In particular, cystic components were detected in 17 out of 27 patients; 8 germinomas and all 9 nongerminomatous GCTs had cysts. The size of the cysts ranged from several millimeters to several centimeters. Multiple cysts occurred in some cases; teratomas and mixed GCT with teratomas were usually honeycomb-like, small to median cystic masses [5]. However, to our knowledge, neither this previous study nor other reports have described cystic GCTs without a solid component.
Here, we report here the case of a suprasellar cystic GCT initially diagnosed as an arachnoid cyst and lacking any solid component until it became a larger cyst with a well-enhanced intracystic mass.
CASE REPORT
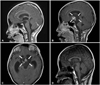
A 10-year-old boy had experienced headache, dizziness, and diplopia for a week before the visit to the emergency room. Brain MRI revealed a suprasellar cyst approximately 2 cm in size and hydrocephalus (Fig. 1A). Based on the impression that the patient had an arachnoid cyst, we gave the patient an infusion of mannitol, and performed endoscopic third ventriculostomy. Tumor markers, such as alpha fetoprotein (AFP) and human chorionic gonadotropin (HCG), were not studied at that time. The ventricular cerebrospinal fluid (CSF) cytology was negative for malignant cells. The pathology findings were an arachnoid membrane with fibrosis and edema, which were features consistent with an arachnoid cyst.
Postoperative brain MRI showed a suprasellar cystic lesion slightly decreased in volume when compared to that of the preoperative MRI. The diplopia and headache were improved. However, the symptoms of polydipsia and nocturia developed postoperatively, suggesting central diabetes insipidus, and 1-deamino-8-D-arginine vasopressin was administered. He remained free of symptoms until the reappearance of headache and dizziness approximately 2 months later. At this time, a 5-6 cm large cystic mass with an internal enhancing component was observed in the suprasellar cistern on brain MRI (Fig. 1B, C). HCG levels were slightly elevated in the serum and the CSF (55 IU/L and 162 IU/L, respectively) but were strikingly elevated in the cystic fluid (14,040 IU/L). AFP levels were not elevated in the serum or the CSF.
Endoscopic third ventriculostomy was followed by craniotomy and tumor removal. Gross total resection of the tumor was performed via a trans-cortical approach. At the time of surgery, the tumor appeared as a grayish cystic lesion composed of a thick arachnoid membrane-like structure just below the foramen of Monro. The thick cystic lesion surrounded a pinkish solid mass. Pathologically, the tumor appeared as a mixed GCT composed of immature teratoma and germinoma. On immunohistological examination, cytokeratin, placental alkaline phosphate, and c-kit were found to be expressed, while AFP and HCG were not detected.
The patient underwent 4 cycles of chemotherapy consisting of carboplatin, etoposide, and bleomycin alternating with cyclophosphamide, etoposide, and bleomycin. MRI after the chemotherapy showed a remarkable decrease in the tumor. The patient received craniospinal irradiation at 30.6 Gy plus local radiation treatment on the residual suprasellar lesion at 23.4 Gy, providing a total dose of 54 Gy. Follow-up MRI showed that a small cystic lesion remained in the suprasellar cistern (Fig. 1D). His serum HCG decreased to normal levels.
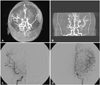
Nine months after the completion of the radiation treatment, the patient developed the clinical features of cerebral ischemia, including sensorimotor paralysis (e.g., left facial numbness and left arm paralysis). The magnetic resonance angiography findings were bilateral T carotid (ICA bifurcation) occlusion with left middle cerebral artery (MCA) occlusion and an apparently focal stenosis at the right P1, which were compatible with moyamoya syndrome (Fig. 2A, B). Single-photon emission computed tomography showed resting hypoperfusion in the right frontal and parietal cortices and decreased vascular reserve in the bilateral MCA territories. A decreased Diamox response was observed in the bilateral frontal, parietal, and temporal cortices. Transfemoral carotid angiography (TFCA) showed severe stenosis in the distal ICAs, MCAs, and ACAs, which are susceptible to moyamoya syndrome (Fig. 2C, D).
Superficial temporal artery-MCA bypass surgery was performed for the treatment of moyamoya syndrome. TFCA after the bypass showed a large subacute infarction involving both cerebellar hemispheres and the brain stem, with swelling in these affected regions. A large, early chronic infarction involving both the entire ACA and MCA territories developed. The patient has been in the intensive care unit for the control of increased intracranial pressure.
DISCUSSION
The diagnosis of intracranial GCT is made by neuroimaging studies in addition to clinical signs and symptoms. Both computed tomography (CT) and MRI are used to detect suprasellar and pineal region masses, but the radiographic characteristics are very similar in all GCTs, therefore limiting their usefulness in determining the exact histology of these tumors. Germinomas can enhance diffusely, while nongerminomatous GCT commonly have associated hemorrhage, causing a more heterogeneous pattern of enhancement [1].
Common suprasellar cyst-appearing lesions include Rathke's cleft cysts, arachnoid cysts, epidermoid cysts, and dermoid cysts. These entities have a specific appearance on CT and MRI and do not demonstrate any enhancement after contrast administration [6]. Arachnoid cysts are CSF covered by arachnoidal cells and collagen that may develop between the surface of the brain and the cranial base or on the arachnoid membrane [7]. Arachnoid cysts occur in approximately 1% of the population. The overwhelming majority of these are found in the supratentorial compartment and, of these, roughly 9-15% occur in the suprasellar region [8]. Arachnoid cysts are a congenital disorder, and most cases begin during infancy; however, onset may be delayed until adolescence [7]. In the present patient, an asymptomatic arachnoid cyst may have existed prior to the diagnosis of GCT. However, the headache, dizziness, and diplopia appeared just one week prior to the visit to the emergency room, and the central diabetes insipidus developed after the improvement of increased intracranial pressure following the first surgery. This rapidly progressive course suggests the suprasellar cystic lesion was a manifestation of GCT rather than a preexisting congenital lesion.
Treatment of intracranial GCT includes a combination of chemotherapy and radiation treatment [2]. Cranial irradiation is associated with adverse effects, including neurocognitive deficits, endocrine abnormalities, secondary neoplasms, and radiation-induced vasculopathy [9]. Progressive intracranial occlusion of the arterial circulation, including moyamoya syndrome, has been a well-documented, uncommon late effect of radiation treatment. Moyamoya syndrome is characterized by the appearance of abnormal collateral vascular networks, which arise adjacent to spontaneously occluded vessels of the circle of Willis. Patients who received radiation treatment to the parasellar region at a young age (<5 years) are the most susceptible to moyamoya disease [10]. The incidence for moyamoya syndrome continues to increase with time, with half of cases occurring within 4 years and 95% of cases within 12 years (median 40 months, range 4-240 months) after radiation treatment [10]. Higher doses of radiation (>50 Gy) to the circle of Willis and neurofibromatosis type 1 result in an increased risk of developing moyamoya syndrome [10]. In this 10-year-old patient, ischemic symptoms developed 9 months after 54 Gy of radiation treatment. However, he did not show any signs of neurofibromatosis type 1.
In conclusion, GCT can be considered as a rare differential diagnosis in the case of a suprasellar cystic mass. An evaluation of tumor markers and a close follow-up will be necessary. In addition, it should be remembered moyamoya syndrome can develop after treatment of suprasellar GCT. Urgent diagnostic work up for suspicious ischemic symptoms and close monitoring for treatment-related complications are necessary in patients with intracranial GCT.
 XML Download
XML Download