

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Bile acids are converted from cholesterol in the liver by numerous cytochrome P450 enzymes, including cholesterol-7α-hydroxylase (CYP7A1), steroid 7α-hydroxylase (CYP27A1) and sterol-27-hydroxylase (CYP27A1).1 Bile acids have been well known to serve as digestive juice to emulsify lipid and promote lipid absorption in the intestinal tract. After endogenous bile acid nuclear receptor Farnesoid X Receptor (FXR) has been reported,23 bile acids are now widely accepted as metabolic regulator to control whole body homeostasis, such as glucose/lipid metabolism and energy expenditure. Thus, the discovery of endogenous bile acid nuclear receptor FXR proposes new perspectives to understand molecular mechanisms and physiological roles of bile acids and their receptors in various tissues to maintain whole body homeostasis.
1. FXR: Nuclear bile acid receptor
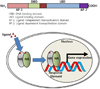
FXR belongs to nuclear receptor superfamily that contains both DNA and ligand binding domains.456 With absence of ligand, nuclear receptors generally bind to their responsive elements with corepressors, including Silencing Mediators of Retinoic acid and Thyroid hormone receptor (SMRT) and histone deacetylases (HDACs) and suppresses their target gene expressions. Upon binding with their ligands, nuclear receptors interact with numerous coactivators and induce their target gene expressions (Fig. 1). FXR heterodimerizes with retinoid X receptor (RXR) and binds to the FXR response element which is localized in the promoter region of FXR target genes.4567 FXR is expressed in various tissues, including liver, adrenal glands, intestine, adipose tissue, endothelial wall, pancreas and kidney.6
Bile acids play as endogenous FXR ligands with high affinity. Both free and conjugated bile acids are able to activate FXR. A well-known hydrophobic bile acid, chenodeoxycholic acid (CDCA) is the most efficacious ligand of FXR (EC50=10µM).289 Generally, the order of potency of bile acids is CDCA>lithocholic acid (LCA)=deoxycholic acid (DCA)>cholic acid (CA).9 Interestingly, hydrophilic bile acids, including ursodeoxycholic acid (UDCA) and muricholic acid (MCA) were not able to activate FXR.10 A synthetic bile acid derivative, 6α-ethylchenodeoxycholic acid (6-ECDCA) has been developed and reported with a higher affinity with FXR than the natural bile acids.9 6-ECDCA recently has been renamed as obeticholic acid, which has recently been approved as a therapeutic reagent for primary biliary cirrhosis (PBC) by Food and Drug Administration,1112 suggesting that FXR is a therapeutic potential target for other metabolic diseases.
2. Regulation of entero-hepatic bile acid circulation
Bile acids are generally conjugated to taurine or glycine upon synthesis. After conjugation, bile acids are secreted into the bile canaliculi. Upon food intake, feeding signal stimulates bile acid secretion into small intestine. Mostly, 95% of secreted bile acids are recycled by entero-hepatic circulation; The remaining 5% of the bile acids excluded in each entero-hepatic circulation. Therefore, the conversion of bile acid from cholesterol compensates an equivalent amount of bile acids to maintain a constant bile acid pool size in the system.
The recycle of 95% of bile acids via entero-hepatic circulation requires the apical sodium-dependent bile salt transporter (ASBT).681314 After transporting bile acids inside enterocytes by ASBT, bile acids are bound by the intestinal bile acid-binding protein (IBABP) which is critical for entero-hepatic circulation of bile acids. Besides ASBT, organic solute transporter alpha and beta (OSTα and β) transport bile acids to the blood vessel, the portal vein.14 Thus, elevated level of bile acids in the intestine induces expression of genes in bile acid transporters to transport bile acids from the intestine to the liver.
3. Physiological roles of FXR in cholesterol/bile acid metabolism
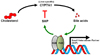
As properties of bile acids are detergent to emulsify lipid substance, accumulation of bile acids in the hepatocytes leads to hepatotoxicity. Bile acid synthesis occurs in the hepatocytes and comprises two pathways: there are two key enzymes, such as CYP7A1 and CYP27A1, respectively.15 Upon FXR activation by bile acids, FXR induces gene expression of small heterodimer partner (SHP), which suppresses gene expression of CYP7A1 to reduce the rate of hepatic bile acid synthesis via negative feedback (Fig. 2).571516
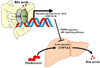
Besides CYP7A1 suppression by SHP, bile acid signaling from intestinal is able to regulate hepatic bile acid synthesis.17 Secretion of bile acids into small intestine activates transcriptional activity of FXR in the intestine, leading to induction of fibroblast growth factor 15/19 (FGF15/19) gene expression as well as bile acid transporter in the small intestine. The elevated FGF15/19 proteins go back to the liver via portal vein and bind to the fibroblast growth factor receptor 4 (FGFR4) of hepatocytes. Upon activation of FGFR4, c-Jun NH(2)-terminal kinase (JNK) signaling pathway in turn suppresses CYP7A1 gene expression to reduce bile acid synthesis in the liver (Fig. 3).18192021
4. Physiological roles of FXR in the hepatic glucose/lipid metabolism
Generation of FXR-null mice has shown impaired glucose tolerance and insulin resistance, suggesting that FXR plays a pivotal role in glucose homeostasis. FXR is able to control gene expression of phosphoenolpyruvate carboxykinase (PEPCK), which is located in the liver to catalyze a crucial step of hepatic gluconeogenesis.62223 Besides PEPCK, FXR activation represses gene expression of glucose-6-phosphatase (G6pc), which catalyzes the hydrolysis of glucose-6-phosphate to glucose. In addition, FXR activation also increases glycogen levels in the liver as via enhancing downstream insulin receptor signaling.623
FXR-null mice studies further revealed the physiological roles of FXR to regulate lipid metabolism.24 Compared with littermate control, FXR-null mice exhibited elevated levels of plasma triglycerides and cholesterol.624 In addition, FXR-null mice also showed higher levels of lipoprotein (HDL) cholesterol in plasma than littermate control, which correlates with a reduction of hepatic expression of scanvenger receptor class B type I (SR-BI), a well-known receptor to clear HDL cholesterol from the blood.2526 Interestingly, transcriptional activation of FXR largely reduced plasma triglyceride levels in wild type mice24; FXR activation is able to suppress gene expressions involved in lipoprotein metabolism including sterol regulatory element-binding protein 1 transcription factor 1c (SREBP-1c), phospholipid transfer protein (PLTP), steroyl-CoA desaturase 1 (SCD-1), the very low density lipoprotein receptor (VLDLR), apolipoprotein C-II, and apolipoprotein E. Altogether, FXR is a key molecule to regulate glucose and lipid homeostasis.
5. Physiological roles of FXR in the intestinal inflammation and anti-tumorigenesis
Previous studies have shown that FXR plays a protective role to control intestinal inflammation to minimize bacterial overgrowth.27 With cholestasis model, bile duct ligation compromised FXR transcriptional activity in the intestine. Interestingly, bile duct ligation led to bacterial overgrowth in the small intestine and translocation into other peripheral tissues.27 FXR activation by synthetic FXR agonist treatment attenuated bacterial overgrowth and reduced intestinal inflammation. Furthermore, FXR has been shown to regulate expression of genes involved in epithelial barriers and antimicrobial peptides, including inducible nitric oxide synthase (iNOS), occluding, and interleukin-18 (IL-18).27 Thus, FXR activation reduces bacterial overgrowth by induction of genes involved in epithelial barriers and antimicrobial peptides.28
Besides intestinal inflammation, FXR has been reported to suppress colorectal tumorigenesis. Though bile acids including deoxycholic acid have been considered as stimulators of tumorigenesis in the colon, activation of FXR by synthetic FXR agonist largely suppressed intestinal tumorigenesis in Apc (Min/+) mice and colitis-associated colorectal cancer model.29 In case of FXR deficiency, Wnt signaling has been upregulated to promote basal proliferative activity in colonic epithelial cells.29 Thus, FXR activation would be useful in the treatment of colon cancer.
6. Physiological roles of FXR in the gut to modulate whole body metabolism
Recent study has shown that beneficial improvements by vertical sleeve gastrectomy (VSG) were diminished in FXR-null mice.30 These results reported that VSG in wild type mice increased circulating bile acids and changed gut microbial communities. Thus, FXR signaling is an important molecular underpinning for the beneficial effects of the VSG.30
In addition to the critical roles of FXR in VSG, a recent study has proposed therapeutic roles of FXR to protect against diet-induced obesity. By using gut-specific FXR synthetic agonist, named Fexaramine, intestinal specific FXR agonism largely increased the thermogenesis and mitochondrial oxidative phosphorylation in brown adipose tissues.31 The intestinal FXR agonism-mediated fat remodeling also led to increased browning of white adipose tissue. Thus, intestinal FXR agonism stimulated energy expenditure in brown and white adipose tissue, leading to increase whole body energy metabolism.31 The molecular mechanisms of how intestinal FXR agonism increased whole body energy metabolism remains still unclear.
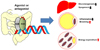
In contrast, another recent study has reported that ablation of intestine-specific FXR led to anti-obesity in obese animal models.32 As previously mentioned, FXR activation in the intestine is able to repress CYP7A1 expression in the liver, resulting in the reduction of bile acid synthesis. Ablation of intestine-specific FXR largely increased hepatic CYP7A1 gene expression and elevated circulating bile acid levels.32 Given that circulating bile acids are able to increase local thyroid signaling in brown adipose tissue and basal energy expenditure,33 intestine-specific FXR ablation resulted in anti-obesity phenotypes in animal models. Furthermore, inhibition of FXR activity in the intestine led to remodeling of gut microbiome to protect against diet-induced obesity.32 Together, both recent studies have suggested and demonstrated that regulation of intestinal FXR transcriptional activity would be a novel therapeutic strategy to protect against metabolic syndromes (Fig. 4).
CONCLUSION
Though bariatric surgery has been well known to improve metabolic parameters in the patients, the molecular mechanisms for beneficial effects still remain unclear. As reported, patients with bariatric surgery usually exhibit elevated circulating bile acid levels, FGF19, and Glucagon-like peptide 1 (GLP-1).343536 Given that bile acids are the endogenous ligand for nuclear receptor FXR, the physiological roles of FXR has been widely accepted as a novel therapeutic target for metabolic syndromes. Though numerous studies have revealed various physiological functions of FXR in the animal models of metabolic diseases, the development of synthetic modulator to regulate FXR transcriptional activities still needs to be required. Since FXR has been reported as a critical molecule to improve beneficial metabolic parameter in VSG using animal model, development of novel synthetic modulator for FXR would be useful therapeutic target for metabolic diseases.
 XML Download
XML Download