

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cardiovascular diseases (CVDs) are the major cause of death in industrialized countries. It is well accepted that aging is the critical risk factor of CVDs, as the prevalence of CVDs is significantly increased in the older population.1 Aging is associated with the progressive deterioration of the structure and function of all types of cells, including vascular endothelial cells (ECs). Indeed, ECs receive cumulative cellular damage during aging, and this age-dependent impairment of the vascular endothelium is a key factor contributing to age-related vascular diseases.2
In order to maintain the integrity of vascular structure and function, the vascular endothelium requires a self-defense system. For example, ECs express endothelial nitric oxide synthase (eNOS) for the generation of NO, which maintains vascular function by inhibiting vasoconstriction, platelet aggregation, leukocyte adhesion, and cell proliferation through a cyclic guanosine monophosphate (cGMP)-dependent intracellular signaling pathway.3 Vascular endothelial growth factor (VEGF) is a representative pro-angiogenic factor, but it also acts as a survival factor for ECs by increasing the synthesis of NO via the phosphorylation of eNOS as well as by inducing the expression of survivin.4 Importantly, endothelium-specific knockout of the VEGF gene results in multiple vascular damages, thus indicating the essential role of endogenous VEGF synthesized by ECs in the maintenance of vascular health.5
ISOLATION OF VASOHIBINS
Many angiogenesis regulators are extrinsic to ECs, but ECs may produce certain angiogenesis regulators by themselves. To search for such novel angiogenesis regulators expressed in ECs, we stimulated ECs in culture with VEGF and then performed DNA microarray analysis to characterize VEGF-inducible genes in these cells.8 Our attention was focused on genes whose functions were undefined. Recombinant proteins of such genes were made and applied to functional bioassays of angiogenesis. This attempt led to the isolation of a gene having anti-angiogenic activity, which we designated as VASH1.6 By subsequent searching in a database, a gene homologous to VASH1 was found and designated as vasohibin-2 (VASH2).9 The overall homology between human VASH1 and human VASH2 is 52.5% at the level of the amino-acid sequence. Moreover, an additional database search revealed that lower organisms such as sea squirt possess one common ancestral vasohibin gene whereas vertebrates have VASH1 and VASH2. Thus, the common ancestral gene seems to have divided into VASH1 and VASH2 during the evolution to the vertebrates.10
VASH1 FOR THE STRESS TOLERANCE OF ECs
Various cellular stresses cause damage to cells, resulting in premature cell senescence and ultimate cell death. When subjected to DNA damage by cellular stresses, the cell cycle is generally arrested, leading to premature senescence. Whereas replicative senescence is the result of telomere shortening, premature cell senescence does not require such an event. Nonetheless, replicative senescent cells are more prone to DNA damage and the resultant premature senescence. A premature senescent cell shows a flattened morphology and will no longer replicate, but it remains metabolically active and exhibits the so-called senescence-associated secretory phenotype.11 One of the major stresses that induce premature cell senescence is oxidative stress, which arises when the balance between production and removal of reactive oxygen species (ROS) favors the pro-oxidation arm. Cardiovascular risk factors such as hypercholesterolemia, hypertension, and diabetes mellitus enhance ROS generation, resulting in oxidative stress; and the oxidative stress-induced damage to the EC is thought to play the most important role in the pathogenesis of vascular diseases.12
VASH1 is expressed in ECs at the site of angiogenesis,6 and terminates angiogenic response.13 However, besides its emergence there on the vascular wall for angiogenesis inhibition, the VASH1 protein is also immunohistochemically detectable especially in arterial ECs under normal conditions.9 The significance of this basal expression of VASH1 was examined by the knockdown of VASH1, which treatment significantly increases the number of premature senescent ECs under basal culture conditions.7 Those senescent ECs are defective in the formation of cell-cell junctions,14 and more susceptible to cell death when exposed to H2O2 or serum deprivation.7 In contrast, when VASH1 is overexpressed in cultured ECs, such cells become resistant to both premature cell senescence and cell death induced by the exposure to H2O2 or serum deprivation. Although the precise mechanism of VASH1-mediated angiogenesis inhibition is not presented at the moment, VASH1 is not an antagonist of VEGF signaling. Thus, the blockage of VEGF signaling causes EC death; and the over-expression of VASH1 rescues ECs from this fate.7 These roles of VASH1 in stress tolerance were confirmed under in vivo conditions by the treatment of mice with paraquat,7 which generates ROS and causes acute organ damage including that to the lungs. When compared with wild-type mice, VASH1 (+/−) mice died in greater numbers due to acute lung injury caused by paraquat. Furthermore, treatment of such mice with an adenovirus vector encoding human VASH1 saved them from death due to paraquat-induced acute lung injury.7 Therefore, VASH1 expressed in the vascular endothelium is active there to protect the vasculature and associated organs from damage.
TARGETS OF VASH1 FOR THE STRESS TOLERANCE OF ECs
1. Superoxide dismutase (SOD) 2
Oxidative stress occurs from an imbalance between the production of ROS and the capacity of antioxidant defense systems to detoxify them. The vascular wall contains enzymes such as SOD, catalase, glutathione peroxidase, heme oxygenase, thoredoxin, and glutathione-S-transferase that act as the antioxidant defense system.15 Among them, the SOD family forms the major antioxidant defense; and SOD exists in 3 isoforms: SOD1 (cytosolic Cu/Zn-SOD), as a soluble enzyme located in the cytoplasm; SOD2 (Mn-SOD), found in the mitochondrial matrix; and SOD3 (extracellular Cu/Zn-SOD), as a secreted enzyme.16 These 3 SODs bind to the superoxide byproducts of oxidative phosphorylation and convert them to hydrogen peroxide and diatomic oxygen, thereby serving a key antioxidant function. SOD2, a 96-kDa homotetramer, is directed to mitochondria by a signal peptide; and because of its localization in mitochondria, SOD2 plays a critical role in protection against oxidative stress.16 ECs are known to express a high level of SOD2,29 which enzyme is thought to play a principal role in protecting the vascular system from oxidative stress generated by various pathophysiological processes.18
It was recently revealed that the knockdown of VASH1 in ECs results in an increase in ROS content and that, moreover, among various antioxidant enzymes, SOD2 is selectively decreased in expression. Alternatively, when VASH1 is overexpressed in ECs, there are an increase in SOD2 expression and a decrease in ROS content in these cells.7 Thus, SOD2 is regarded as the target of VASH1 that reduces oxidative stress in ECs.
2. Sirtuin1 (SIRT1)
Additional target of VASH1 that governs the antioxidant defense of ECs is SIRT1. This enzyme is a member of the mammalian NAD+-dependent deacetylase and ADP-ribosyltransferase family, which is orthologous to Silencing information regulator 2 (Sir2).19 Sir2 was originally isolated from yeast as a chromatin-silencing component that plays diverse biological roles in longevity, genome stability, and cellular metabolism.20 Mammalian sirtuins have 7 isoforms (SIRT1-7). SIRT1-3 and SIRT5-7 catalyze NAD+-dependent substrate-specific deacetylation; whereas SIRT4 has NAD+-dependent mono-ADP-ribosyltransferase activity, and SIRT6 possesses both deacetylase and auto-ADP-ribosyltransferase activities.21 Among them, SIRT1 is most closely related to Sir2, and is now considered to be responsible for the stress resistance of cells. In particular, a number of reports indicate that vascular SIRT1 protects vessels from vascular diseases including atherosclerosis and diabetic vascular complications.22232425
It was recently revealed that the knockdown of VASH1 in ECs results in a decreased SIRT1 protein level as well as in reduced activity and that, in contrast, when VASH1 is overexpressed in ECs, there is an increase in the level of SIRT1 protein.7 Moreover, when this increase in SIRT1 protein is specifically knocked-down, the stress-resistant effect of VASH1 on ECs disappears.7 Thus, SIRT1 is also regarded as a target of VASH1 that increases the stress tolerance of ECs.
POST-TRANSCRIPTIONAL REGULATION OF VASH1 SYNTHESIS
Gene expression begins with the transcription of DNA for the generation of mRNA and ends with its translation to protein. Post-transcriptional regulation controls the fate of mRNAs at the steps of splicing, export, stabilization, and translation; and these processes are regulated by the interaction of cis-regulatory elements on mRNA and trans-acting factors such as RNA binding proteins (RBPs) and microRNAs that bind to these elements.
1. HuR for stress-induced VASH1 synthesis in ECs
When ECs are exposed to pro-angiogenic stimuli such as VEGF, ECs gradually increase their expression of VASH1 mRNA and VASH1 protein synthesis over a 24-hour period.6 This VEGF-mediated increase of VASH1 mRNA is not influenced by the actinnomycin D treatment indicating that this increase of VASH1 mRNA is mediated by the transcriptional activation.26 However, when ECs are exposed to cellular stresses, ECs promptly increase their level of VASH1 protein within 3 hours, but this increase is not accompanied by increased transcription of VASH1 mRNA. This prompt increase in VASH1 protein synthesis in ECs is responsible for stress tolerance, and is mediated by an RNA binding protein (RBP), namely, HuR, that acts in a post-transcriptional manner.7
HuR belongs to the embryonic lethal abnormal vision (ELAV) family of RBPs that bind to U-rich and/or AU-rich elements in the 3′-UTRs of their target mRNAs, and it prevents their degradation and enhances translation.27 There are 4 members of the ELAV protein family, i.e, HuB, HuC, HuD, and HuR. Whereas HuB, HuC, and HuD are selectively expressed in the nervous system and play roles in neuronal differentiation and plasticity, HuR is ubiquitously expressed and involved in numerous cellular responses.27 HuR is present predominantly in the nucleus under normal conditions. When cells are exposed to cellular stresses, HuR is translocated to the cytoplasm, where it interacts with mRNAs and elicits cellular stress responses.28
2. MicroRNA for downregulation of VASH1 with aging
MicroRNAs are small non-coding RNAs, which regulate gene expression at the post-transcriptional level via target mRNA degradation and/or translational suppression via binding to the 3′-UTR.29 It is estimated that approximately one third of all animal genes are controlled by microRNAs, and thus microRNAs exhibit a wide range of biological processes including aging.30 Indeed, increasing evidences suggest that changes in the expression profile of microRNAs contribute to cellular senescence, aging and aging-related diseases. Vascular aging is associated with endothelial cell dysfunction, and endothelial cell dysfunction is thought to be critical for the development of various vascular diseases,31 and several studies were directed to examine the relationship between certain microRNAs such as miR-17, miR-18, miR-19, miR-22 and miR-34 and cardiovascular aging.32
The expression of VASH1 is found to be decreases with replicative senescence of ECs.33 To explore the mechanism of this down-regulation of VASH1 in senescent ECs, the expression of microRNAs between old and young Human umbilical venous endothelial cells (HUVECs) is compared by performing microarray analysis. Among the top 20 microRNAs that are expressed at a higher level in old HUVECs, the third highest microRNA, namely, miR-22-3p, has its binding site on the 3′-UTR of VASH1 mRNA. Experiments with microRNA mimic and anti-miR have revealed that miR-22-3p is involved in the down-regulation of VASH1 in ECs during replicative senescence.33 MiR-22 is previously reported to promote senescence of endothelial progenitor cells by targeting AKT3.25 The report by Takada et al. adds VASH1 to the list of miR-22 targets that are down-regulated with replicative senescence of ECs, and that might be responsive to aging-associated vascular diseases.
PATHOPHYSIOLOGY OF VASH1 IN VASCULAR DISEASES
1. Defensive role in the vascular wall
Because of more serious paraquat-induced acute organ damage in VASH1 (+/−) mice than in their wild-type littermates,7 the role of endogenous VASH1 in vascular diseases was further clarified by comparing the development of STZ-induced diabetic vascular disease in VASH1 (+/−) mice with that in their wild-type littermates.34 The results indicated that albuminuria is significantly exacerbated in the diabetic VASH1 (+/−) mice when compared with that in their diabetic wild-type littermates. This disorder was found to be associated with glomerular basement membrane thickening and a reduction in slit diaphragm density. Furthermore, the renal hypertrophy and glomerular accumulation of mesangial matrix and type IV collagen are exacerbated in the diabetic VASH1 (+/−) mice compared with these features in their diabetic wild-type littermates.
Alternatively, when VASH1 is overexpressed in the lungs of VASH1 (+/−) mice by the intra-tracheal administration of adenoviral vectors encoding human VASH1 (AdhVASH1), the paraquat-induced acute lung injury is almost completely prevented; and, in addition, this maneuver increases the expression of SOD2 and SIRT1 in the lungs.7 Such a protective activity of VASH1 was further documented in murine diabetes models, as human VASH1 delivered from the liver infected with AdhVASH1 exhibits therapeutic effects on diabetic renal alterations.3536 These loss-of-function and gain-of-function studies have clearly demonstrated the defensive role of VASH1 in the vascular system.
CONCLUDING REMARKS
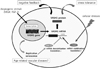
This mini review summarized a novel molecular basis for vascular health regulated by VASH1. VASH1 is a negative-feedback regulator of angiogenesis synthesized by ECs, which also increases stress tolerance of ECs via the induction of SOD2 and SIRT1. Besides its transcriptional induction by angiogenic stimuli, the synthesis of VASH1 receives post-transcriptional regulation. Cellular stresses increase the synthesis of VASH1 via HuR, whereas replicative senescence of ECs decreases the synthesis of VASH1 via miR-22. Such senescence-related decrease of VASH1 synthesis might be deteriorus for vascular health (Fig. 1).
 XML Download
XML Download