

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Systemic lupus erythematosus (SLE) is a chronic systemic autoimmune disease. Acute myocardial infarction (AMI) is an uncommon initial presentation for systemic lupus erythematosus. The incidence of myocardial infarction is 5 times as high in patients with lupus as in the general population, and in young women the age-specific incidence is increased by a factor of as much as 50.1 There are several proposed mechanisms for AMI in lupus patients, including traditional risk factors and nontraditional risk factors. We report a case of AMI in a young man with SLE who developed recurrent AMI and received percutaneous coronary intervention (PCI) and also aim to discuss the mechanisms.
CASE REPORT
A 37-year-old man with a 12-year history of SLE had been treated with a corticosteroid since being diagnosed as having SLE at the age of 25 years. He had history of hypertension, hyperlipidemia and current smoking.
In August 2005, at the age of 30 years, the patient was admitted to the hospital with chest pain of 12-hour duration. Physical examination showed normal results and no murmur or rales were auscultated. His initial electrocardiogram (ECG) showed normal sinus rhythm and T-wave inversion in leads II, III, aVF. An emergent echocardiogram demonstrated hypokinesia of the inferior walls. On the laboratory studies, the white blood count was 4.6×103/µL, the hemoglobin was 14.2 g/dL, and the platelets were 169×103/µL. The erythrocyte sedimentation rate (ESR) was 19 mm/hr (normal 0 to 20) and the C-reactive protein (CRP) concentration was 0.1 mg/L (normal 0 to 0.3). The initial coagulation studies showed a partial thromboplastin time of 39.4 seconds (normal 26.5 to 41.1) and an international normalized ratio of 1.02 (normal 0 to 1.2). The peak level of creatine kinase (CK) was 2,347 U/L (normal 35 to 172), CK-MB 95.4 U/L (normal 2.3 to 9.5) and troponin-I 19.0 ng/mL (normal 0 to 0.05). The antinuclear antibody test was positive at 1:160 with a homogeneous pattern. The double-stranded DNA was negative and the antiphospholipid antibody (APLA) panel was negative. The levels of complement were low, with the levels of Complement (C) 3 at 58 mg/dL (normal 90 to 180) and C4 at 9.31 mg/dL (normal 10 to 40). The lipid profile showed total cholesterol (TC) 125 mg/dL (normal 150 to 200), triglycerides (TG) 89 mg/dL (normal 0 to 200), high density lipoprotein-cholesterol (HDL-C) 26.1 mg/dL (normal 40 to 80), and low-density lipoprotein-cholesterol (LDL-C) 88 mg/dL (normal 0 to 120).
Coronary angiogram (CAG) revealed huge aneurysmal dilatations of the three proximal coronary arteries with slow Thrombolysis in the myocardial infarction (TIMI) flow (TIMI 2 antegrade flow). An intravascular ultrasound (IVUS) revealed huge anerysmal dilatations with thrombi in all three coronary arteries. The patient received aggressive anti-thrombotic medical therapy including the standard regimen of aspirin, clopidogrel and heparin without PCI. He had good recovery and there were no cardiac events during clinical follow-up. Follow-up CAG was performed at 8-months after discharge and it revealed good distal flow and huge multiple aneurysms in the three coronary arteries, but without thrombus.
In February 2013, at the age of 37 years, the patient suffered from severe chest pain and presented to our hospital again. On admission, physical examination showed blood pressure was 120/80 mmHg and heart rate was 70 beats/min, and no murmur or rales were detected. The ECG showed normal sinus rhythm and T-wave inversion in leads III, aVF and V5-V6 (Fig. 1). The echocardiogram showed good left ventricular systolic function and akinesia in the inferior walls. The white blood count was 9.6×103/µL, the hemoglobin was 14.5 g/dL and the platelets were 204×103/µL. The ESR was 22 mm/hr and the CRP concentration was 0.19 mg/L. The cardiac enzymes were elevated, with CK of 253 U/L, CK-MB of 104.2 U/L, and troponin I of 12.9 ng/ml. The levels of C3 at 99.2 mg/dL and C4 at 19.1 mg/dL. The lipid profile was favorable with levels of TC 154 mg/dL, TG 193 mg/dL, HDL-C 38 mg/dL, and LDL-C 97 mg/dL.
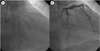
CAG revealed total occlusion in the distal left circumflex artery (LCX), and more aggravated stenosis in the middle left anterior descending coronary artery (Fig. 2). After pre-dilation with a 1.0×5 mm balloon and the residual stenosis is 75%. So 3.0×19 mm bare-metal stent (Coroflex stent, B. Broun, Germany) was implanted (Fig. 3A). The final angiogram showed TIMI 3 flow without residual stenosis (Fig. 3).
He was discharged 1 week after presentation, and with aspirin, clopidogrel, bisoprorlol, candesartan, and atorvastatin. At 1-month follow-up visit, he did not complain of any recurrence of chest pain.
DISCUSSION
SLE is a chronic and multi-systemic autoimmune disorder occurring predominantly in women. SLE has many clinical manifestations, one of which is coronary artery disease including AMI. The most common cause of death in SLE patients affected by disease for more than 5 years is cardiovascular disease.2 Coronary artery disease (CAD) is one of the cardiovascular manifestations observed in young SLE patients. The clinical manifestations of CAD in SLE can result from several pathophysiologic mechanisms, including atherosclerosis, arteritis, thrombosis, embolization, spasm, and abnormal coronary flow.3,4 Patients with SLE have increased risks of AMI. Recent case-control series have confirmed that the risk of myocardial infarction in patients with SLE is increased between 9- and 50-fold over that in the general population.4-6
Korkmaz et al.,7 in a review of the English literature from 1975 to 2006, found records of 50 SLE patients with AMI under 35 years old. According to the characteristics of their coronary lesions, the 50 patients were divided into 3 groups: 16 with normal coronary arteries or coronary thrombosis (group I), 12 with coronary aneurysm or arteritis (group II), and 22 with coronary atherosclerosis (group III). Five patients in group I showed normal coronary arteries, and three patients exhibited anti-cardiolipin antibodies and/or lupus anticoagulant; the other 11 patients showed isolated coronary thrombosis. Most patients in the group I (93%) were positive for Antiphospholipid antivodies (APLAs), but among the 22 patients in group III, only one patient was positive for APLAs.
In this case, our young patient experienced AMI twice. The major cause of the first AMI may be attributed to coronary artery thrombi within multiple coronary aneurysms. Coronary artery thrombosis is often related to the presence of APLAs in SLE patient. The presence of APLAs predisposes some patients to thrombosis, and it has also has been associated with valvular thickening and nonbacterial endocarditis.8 Coronary aneurysm is rare in SLE and confirmation of etiology is usually made at postmortem examination and likely to result in myocardial infarction. The precise mechanisms of coronary aneurysm in patients with SLE are unknown. The vascular endothelial dysfunction and vascular inflammation may play an important role in coronary aneurysm.9
The major cause of the second AMI may be attributed to coronary artery atherosclerosis and endothelial injury leading to plaque rupture. The young age of many of the patients with lupus, atherosclerosis remains the most common cause of AMI.10 Our patient had traditional risk factors for atherosclerosis, including hypertension, dyslipidemia as well as smoking. However, evidence suggests that traditional risk factors alone do not explain the increase in CAD observed.11,12 The nontraditional CAD risk factors in SLE are Lupus nephritis, presence of proinflammatory cytokines, some of inflammatory mediators, APLAs, anti-oxidative modified low-density lipoproteins antibodies, long-term use of corticosteroid and cumulative dose of glucocorticoids.13
The optimal treatment of AMI from each etiology may be different. For AMI from atherosclerotic disease, the early PCI, angioplasty, and stent placement to achieve revascularization is the best treatment. Coronary vasculitis, on the other hand, may be a specific manifestation of systemic inflammation in SLE, and conventional thrombolysis may fail to achieve reperfusion. Instead, treatment for vasculitis with systemic steroids may be more appropriate. For non-atherosclerosis acute thrombosis, aggressive anti-platelet therapy is the best treatment. For coronary vasospasm treatment with intracoronary nitrate administration can resolve both symptoms of chest pain and angiographic occlusion.14
In conclusion, our patient with SLE developed AMI twice with different causes. We believe that the best treatment of AMI may be different according to its etiology.


 XML Download
XML Download