

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Introduction
Ebola virus causes hemorrhagic fever with mortality rates up to 90%. In a retrospective study of Ebola virus disease-positive patients affected by the 2014 outbreak of Ebola virus in West Africa (Liberia and Sierra Leone), the mortality rates for children under 5 and adults over 55 were found to be 89.1% and 71.4%, respectively [1]. Despite its high mortality rates, there are still neither licensed anti-viral therapeutic drugs nor preventive vaccines against Ebola virus infection. Passive transfer of glycoprotein (GP)-specific antibodies (Abs) prior to or up to 1 hour after viral challenge provides protection against lethal Ebola virus infection in an animal model; transfer of sera specific for GP, rather than those against other viral proteins, confers the most effective protection [23]. The GP epitopes in that study were associated with Ab-mediated protection [2]. In the case of survivors of Ebola virus infection, early pro-inflammatory responses and persistent increases in Ebola virus-specific IgG levels were detectable [4]. Furthermore, 100% of monkeys treated with an Ab cocktail (ZMAB, composed of three different monoclonal Abs [MAbs] that are mouse-human chimeric and specific for GP) within 24 hours following a lethal challenge with Ebola virus were protected from death, while all animals without ZMAB treatment died within 5 days of viral infection [5]. When the surviving animals were re-challenged with lethal Ebola virus doses, they maintained their activated immune status and were protected from the virus infection [6]. Collectively, these data highlight the important function of GP-specific Abs for controlling Ebola virus infection.
Electroporation (EP) has been known to be the most effective DNA delivery method for the induction of adaptive immune responses. For example, during intramuscular (IM)-EP, EP-generated electric pulses induced more pore formation in the cell membranes of the muscle cells and antigen presenting cells (APCs), leading to more efficient entry and expression of DNA vaccines in the cells, and facilitating immune induction [78]. In addition, stimulation caused by both needle injection and EP attracts more immune cells, including APCs and T cells, to the DNA injection site, leading to enhanced induction of adaptive immune responses [910]. Unlike recombinant protein-based vaccines, moreover, DNA vaccines are thought to be better at inducing Abs that recognize the native forms of protein antigens, as DNA vaccines express their encoded proteins in the target cells in a manner similar to natural viral infection. In this context, it is likely that IM-EP of DNA vaccines will efficiently elicit Abs capable of blocking viral infection. In addition, purification of DNA vaccines is far easier than purification of proteins. To our knowledge, DNA vaccines have not been reported for use in generating hybridomas. Thus, we tested whether DNA vaccination could be used for hybridoma production.
In this study, we utilized IM-EP delivery of GP DNA vaccines to generate MAbs. We generated 960 hybridomas via fusion of splenocytes from GP DNA vaccine-immunized mice with myeloma cells; among those hybridomas, 12 displayed reactivity to GP in an enzyme-linked immunosorbent assay (ELISA). Using limiting dilution subcloning, we obtained 12 monoclonal hybridomas, among which 11 secreted IgM types and 1 secreted an IgG type, in particular IgG2a. The 11 purified IgM Abs, but not the IgG, failed to recognize GP in an ELISA. However, boosting with the protein antigen, GP facilitated more efficient generation of IgG-secreting hybridomas than GP DNA vaccination alone. These data suggest that immunization with GP DNA vaccines leads to preferential production of hybridomas secreting IgM Abs with little antigen specificity, and that boosting with GP can overcome this propensity. Taken together, these data suggest that boosting with GP could be useful for producing hybridomas that secrete antigen-specific IgG Abs.
Materials and Methods
Animals and cells
Six-week-old, female BALB/c mice were purchased from Daehan Biolink (Eumseong, Korea). Nude mice of BALB/c origin were purchased from OrientBio (Seongnam, Korea). The mice were cared for under the guidelines of the Kangwon Institutional Animal Care and Use Committee-approved protocols. Sp2/0-Ag14 cells were purchased from Korean Cell Line Bank (Seoul, Korea). The cells were maintained in cDMEM (Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 1% L-glutamine, and 1% penicillin/streptomycin).
Production of recombinant GP and DNA vaccines encoding Ebola virus GP
Ebola virus (Zaire Ebola virus strain Gabon-94) GP genes (2,043 bp, GeneBank No. U77384.1) were codon-optimized and synthesized by GenScript (Hong Kong). The synthesized GP DNA sequence was cloned into a pcDNA3.1 backbone vector (Invitrogen, Carlsbad, CA, USA), which was designated as pcDNA3-GP. Recombinant GP was generated in HEK293 cells and purchased from Y-Biologics (Daejeon, Korea). For the production of GP1a, GP1b, GP1c, GP1d, and GP1e, DNA sequences encoding the 5 GP1 regions were generated using the primer sets in Table 1. Each DNA fragment was digested with EcoRI and XhoI, and then gel-purified. The DNA fragments were then cloned into the EcoRI and XhoI sites of a pET vector (Novagen, Madison, WI, USA). The plasmid constructs were transformed into Escherichia coli DH5α cells, which were selected against kanamycin. The pET-GP1a, pET-GP1b, pET-GP1c, pET-GP1d, and pET-GP1e vectors were purified, transformed into BL21 (DE3) cells, and then incubated in Luria-Bertani broth supplemented with kanamycin at a final concentration of 30 µg/mL. The cells were incubated in a shaker until the absorbance at 600 nm was between 0.6 and 0.8 absorbance units. The proteins were induced by the addition of 1 mM isopropyl-1-thio-β-D-galactopyranoside for 3 hours. The cell pellets were collected at 3,000 ×g for 20 minutes and subjected to a single freeze-thaw cycle at –20℃. Cell pellets were resuspended in 5 mL of 8 M urea buffer (pH 8.0) per gram wet weight. The cells were lysed by stirring for 15–60 minutes at room temperature and centrifuged at 25,000 ×g for 30 minutes. The cell supernatants were collected and passed through a Ni-NTA resin column (Qiagen, Valencia, CA, USA) that had been pre-equilibrated with 8 M urea buffer (pH 8.0). The resin was washed with 5 volumes of Buffer B (8 M urea buffer [pH 8.0]) and then with 5–10 volumes of Buffer C (8 M urea buffer [pH 6.3]). In the final step, His-tagged GP1a, GP1b, GP1c, GP1d, and GP1e were eluted with 10 mL of Buffer C containing 200 mM imidazole. The protein solutions were then dialyzed in 6 M urea buffers, followed by 4 M urea buffers, at 2-hour intervals. This was followed by overnight dialysis in phosphate-buffered saline (PBS). The protein concentration was determined using the bicinchoninic acid protein assay kit (Thermo Scientific, Rockford, IL, USA).
Delivery of GP DNA vaccines and GP
For IM-EP delivery of DNA vaccines, mice were injected intramuscularly with 50 µg of pcDNA3-GP per mouse in a final volume of 50 µL PBS using a 31-gauge needle. The injections were followed by EP at 0.2 V for 4 seconds using a Cellectra system (VGX International Inc./Inovio), in accordance with the manufacturer's protocol. Plasmid DNA was produced in bacteria and purified using endotoxin-free Qiagen kits according to the manufacturer's protocol (Qiagen). For the delivery of GP, mice were injected intravenously with 20 µg GP per mouse in a final volume of 200 µL PBS.
Production of hybridomas with GP-binding activity
Mice were sacrificed at the indicated time points following their last injection, and their splenocytes were prepared for fusion. The fusion was performed as previously described [11]. In brief, the splenocytes (7×107 cells) were fused with 2×107 Sp2/0-Ag14 cells using 50% polyethylene glycol 4000 (PEG 4000). The cells were incubated at 37℃ for 10 days in cDMEM containing hypoxanthine-aminopterin-thymidine (HAT). Then, the cells were incubated with cDMEM containing hypoxanthine-thymidine (HT). After 15 days, the cell supernatants were screened to detect the presence of Ab-secreting hybridomas using ELISA. For the production of MAbs, the Ab-secreting hybridomas were diluted to less than a single cell per well using the limiting dilution subcloning method. PEG 4000, HAT, and HT were purchased from Sigma-Aldrich (St. Louis, MO).
Purification of MAbs
Hybridoma cells were cultured in cDMEM containing HT. The cell supernatants were collected and used for the purification of MAbs. Alternatively, ascites fluids were generated by injecting 1×106 hybridoma cells intraperitoneally into nude mice. The nude mice were injected intraperitoneally with 500 µL of pristane 2 weeks prior to hybridoma cell injection. Nude mice, which lack T cells, were chosen as they tended to form ascites fluid more easily than BALB/c mice. When the mice became noticeably large, we collected ascites fluids with an 18-gauge needle attached to a 5-mL syringe. After incubating the fluids at 37℃ for 1 hour and then overnight at 4℃, we centrifuged them at 3,000 ×g for 10 minutes. We removed an oil layer and the cell pellets, and collected the fluid supernatants, which were used for the purification of MAbs. MAbs (IgG) were purified using a protein G resin column in accordance with the manufacturer's protocol (Peptron, Daejeon, Korea). Briefly, the protein G resin column was pre-equilibrated with 10 mL of binding buffer (0.1 M sodium phosphate buffer with 0.15 M NaCl, pH 7.4). Samples were filtered through a 0.2-µm filter and mixed with an equal volume of binding buffer. The mixed samples were loaded onto the resin column and washed twice with 10 mL of binding buffer. Finally, the bound Abs were eluted by adding elution buffer (0.2 M glycine buffer, pH 2.5). In particular, we added 65 µL of neutralization buffer (1 M Tris/HCl buffer, pH 9.0) to 0.5 mL of each eluted sample fraction to adjust the pH. We utilized 2 different methods to purify IgM. In the first method, IgM was purified using HiTrap IgM purification columns in accordance with the manufacturer's protocol (GE Healthcare, Cleveland, OH, USA). In brief, the columns were pre-equilibrated with 10 mL of binding buffer (20 mM sodium phosphate and 0.8 M (NH4)2SO4, pH 7.5). Samples were passed through a 0.2 µm filter and mixed with an equal volume of binding buffer. The mixed samples were loaded onto the column, followed by washes with 15 mL of binding buffer. The bound IgM was eluted by adding elution buffer (20 mM sodium phosphate, pH 7.5). In the second method, IgM was purified using a protein L resin column in accordance with the manufacturer's protocol (Amicogen, Jinju, Korea). Briefly, the protein L resin column was pre-equilibrated with 10 mL of binding buffer (0.1 M sodium phosphate buffer with 0.15 M NaCl, pH 7.4). Samples were passed through a 0.2-µm filter and mixed with an equal volume of binding buffer. The mixed samples were loaded onto the resin column and washed twice with 10 mL of binding buffer. Finally, the bound Abs were eluted by adding elution buffer (0.2 M glycine buffer, pH 2.5). In particular, we added 65 µL of neutralization buffer (1 M Tris/HCl buffer, pH 9.0) to 0.5 mL of each eluted sample fraction to adjust the pH. Finally, the Abs were dialyzed in PBS.
ELISA
Each well of a 96 well plate was coated at 4℃ overnight with recombinant GP (0.5 µg/mL in PBS). The next day, the plate was washed 3 times with washing buffer (PBS+0.05% Tween 20) and blocked for 1 hour with blocking buffer (washing buffer +2% bovine serum albumin). Then, the plate was reacted for 1 hour with horseradish peroxidase (HRP)–conjugated anti-mouse IgG (H+L), HRP-conjugated anti-mouse IgM, HRP-conjugated anti-mouse IgA, or HRP-conjugated anti-mouse IgG (Fc-specific) diluted in blocking buffer. After washing the plate, an ABTS solution (Merck Millipore, Billerica, MA, USA) was added to each well. The optical density was read at 405 nm using an ELISA reader. For the determination of the relative levels of GP-specific IgG subclasses, we substituted HRP-conjugated anti-murine IgG1, IgG2a, IgG2b, and IgG3 Abs (Thermo Scientific) for HRP-conjugated anti-murine IgG (H+L). HRP-conjugated anti-mouse IgG (H+L) and HRP-conjugated anti-mouse IgM were purchased from Komabiotech (Seoul, Korea). HRP-conjugated anti-mouse IgG (Fc-specific) and HRP-conjugated anti-mouse IgA were purchased from Sigma-Aldrich and Thermo Scientific, respectively.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and Western blot assay
Protein samples were separated on 12%–15% sodium dodecyl sulfate (SDS)–polyacrylamide gels. The proteins on the gels were analyzed by staining with Brilliant Blue R250 staining solution (Elpis-Biotech, Daejeon, Korea). The proteins were also electrophoretically transferred to Immobilon-P membranes (Merck Millipore). The membranes were pre-equilibrated with TBST solution ([10 mM Tris-HCl [pH 8.0], 150 mM NaCl, and 0.1% Tween 20] containing 5% skim milk for 1 hour. For the detection of Abs, the membranes were incubated with HRP-conjugated anti-mouse IgG (H+L) (Komabiotech) for 1 hour at room temperature. In each step, the membrane was washed 3 times with TBST. The immunoreactive protein bands were visualized using ECL detection reagents (PhileKorea, Seoul, Korea).
Results
Production of Ebola virus GP DNA vaccines, and induction of GP-specific Ab responses by GP DNA vaccination
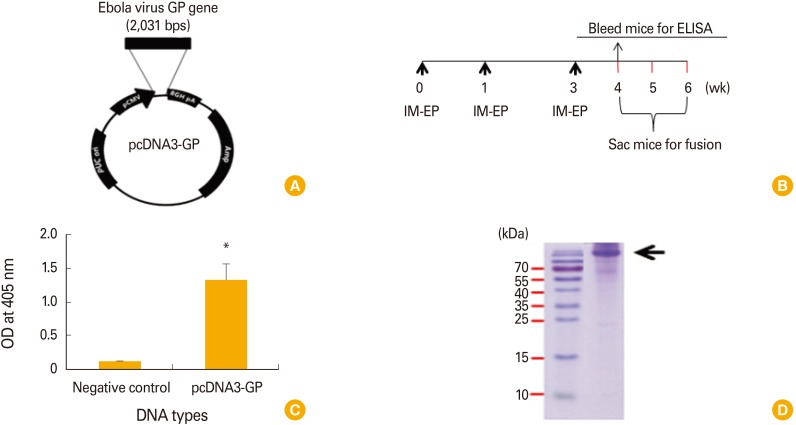
First, we constructed DNA vaccines coding for Ebola virus GP using the codon-optimized Ebola virus GP DNA sequences, which were designated as pcDNA3-GP (Fig. 1A). To test whether GP DNA vaccines could induce antigen-specific Ab responses, mice were immunized by IM-EP with pcDNA3-GP at 0, 1, and 3 weeks and bled at 4 weeks (Fig. 1B). Fig. 1C shows the ELISA data for the GP DNA vaccines. The GP DNA vaccines induced significantly greater antigen-specific Ab responses than a control pcDNA3 vector. Recombinant GP (140 kDa) was used as a coating antigen for ELISA (Fig. 1D). This result suggests that GP DNA vaccines can elicit antigen-specific Ab responses in vivo.
Production of hybridomas secreting Abs specific for Ebola virus GP
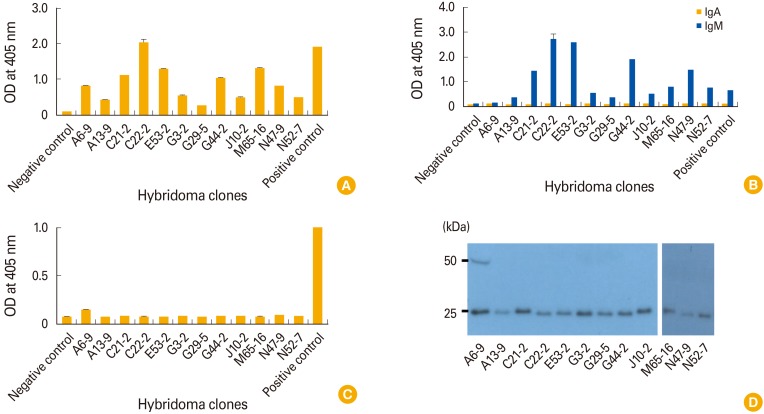
To obtain splenocytes for fusion, mice were injected by IM-EP with GP DNA vaccines. One week after the last injection, the mice were sacrificed and their splenocytes were used for fusion, as in Fig. 1B. In this assay, we generated 162 hybridomas, among which five were found to secrete Abs reactive to GP in an ELISA using HRP-conjugated anti-mouse IgG (H+L) as the secondary Ab. We utilized hybridoma cell supernatants to select hybridomas secreting GP-specific Abs. The five hybridomas were designated as A6, A13, B48, C21, C22, and D3. We also generated 312 hybridomas from the mice sacrificed at 2 weeks after the last injection (as in Fig. 1B), among which four were found by ELISA to secrete Abs reactive to GP. These four hybridomas were designated as E53, G3, G29, and G44. In addition, we produced 578 hybridomas from the mice sacrificed at 3 weeks after the last injection (as in Fig. 1B), among which four were found by ELISA to secrete Abs reactive to GP. The four hybridomas were designated as J10, M65, N47, and N52. For MAb production, the 13 hybridomas were diluted to generate single cell clones, using the limiting dilution subcloning method. As a result, 13 monoclonal hybridoma clones were obtained. However, clone D3-11 became contaminated. Thus, 12 clones were selected and designated as A6-9, A13-9, C21-2, C22-2, E53-2, G3-2, G29-5, G44-2, J10-2, M65-16, N47-9, and N52-7. Next, we tested by ELISA if the cell supernatants from the 12 clones contained MAbs that could bind to GP. As seen in Fig. 2A, all 12 clones displayed reactivity to GP in an ELISA using HRP-conjugated anti-IgG (H+L) as the secondary Ab. However, only clone A6-9 failed to react with GP in an ELISA using HRP-conjugated anti-IgM as the secondary Ab (Fig. 2B); the other 11 displayed reactivity with anti-IgM. Moreover, none of the 12 clones showed reactivity to GP in an ELISA using HRP-conjugated anti-IgA as the secondary Ab (Fig. 2B). Only the cell supernatant from clone A6-9 showed reactivity to GP in an ELISA using HRP-conjugated anti-IgG (Fc-specific) as the secondary Ab (Fig. 2C). These data suggest that 11 of the clones may produce antigen-specific IgM, with the exception of clone A6-9, which appeared to secrete antigen-specific IgG. To confirm this finding, cell supernatants from the 12 clones were tested by Western blot assay using HRP-conjugated anti-IgG (H+L). As seen in Fig. 2D, samples from clone A6-9 showed the presence of both IgG H (50 kDa) and L (25 kDa) chains, whereas those from the remaining 11 clones displayed only L chains. This result suggested that 11 of the clones secreted antigen-specific IgM, but clone A6-9 secreted antigen-specific IgG. Taken together, these results indicate that IgM-secreting hybridomas are predominantly generated by immunization with DNA vaccines coding for Ebola virus GP.
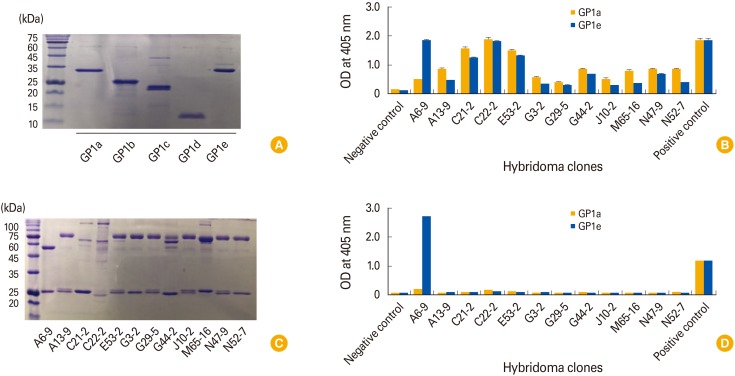
Production of Ebola virus GP1a, GP1b, GP1c, GP1d, and GP1e, and assessment of the capacity of the 12 MAbs to recognize GP1a and GP1e
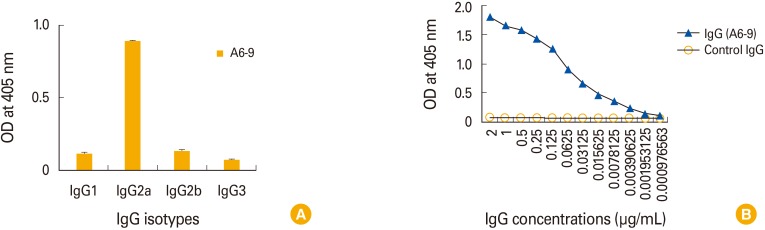
A whole Ebola virus GP, including signal peptides, the putative receptor-binding domains, mucin-like region and transmembrane region, is shown in Fig. 3. We generated four recombinant GP1 constructs spanning the receptor-binding domains and one recombinant GP1 spanning the mucin-like region. The amino acid sequence numbers of the five recombinant GP1 constructs (GP1a, GP1b, GP1c, GP1d, and GP1e) are also shown in Fig. 3. The recombinant GP1a, GP1b, GP1c, GP1d, and GP1e molecules were generated using the E. coli system. Purified GP1a, GP1b, GP1c, GP1d, and GP1e ran on an SDS-polyacrylamide gel at 32 kDa (28-kDa GP1a protein plus 4-kDa His-tag), 24 kDa (20-kDa GP1b protein plus 4-kDa His-tag), 20 kDa (16-kDa GP1c protein plus 4-kDa His-tag), 11 kDa (7-kDa GP1d protein plus 4-kDa His-tag), and 35 kDa (24-kDa GP1e protein plus 4-kDa His-tag), respectively (Fig. 4A). In particular, the molecular mass of GP1e was 7 kDa larger than predicted. The concentrations of GP1a, GP1b, GP1c, GP1d, and GP1e were calculated as 0.78, 0.3, 4.5, 3.1, and 1.6 mg/mL, respectively. Next, we evaluated the reactivity of the cell supernatants from the 12 clones to GP1a and GP1e in ELISA, using HRP-conjugated anti-IgG (H+L) as the secondary Ab. As shown in Fig. 4B, the cell supernatants from the 11 clones displayed higher, though variable, binding reactivity to both GP1a and GP1e than to the negative control. In contrast, cell supernatants from clone A6-9 displayed binding activity to GP1e, but little to GP1a. These data suggest that 11 of the hybridoma clones may secrete IgM Abs with high avidity to multiple antigens. Therefore, we purified MAbs from the cell supernatants or the ascites fluids of the 12 clones. We used a protein G resin column to purify IgG from the cell supernatant of clone A6-9. IgM was purified either from ascites fluids for clones C21-2, C22-2, J10-2, and M65-26 using the HiTrap IgM purification kit, or from the cell supernatants of clones A13-9, E53-2, G3-2, G29-5, J10-2, N47-9, and N52-7 using a protein L resin column, based on the availability of the HiTrap IgM purification kit or L resin column. The purified IgG from clone A6-9 displayed both a 50-kDa H and 25-kDa L chain on an SDS-polyacrylamide gel, whereas the 11 purified IgM Abs displayed 70- to 75-kDa H and 25-kDa L chains (Fig. 4C). Next, we tested the abilities of the purified IgG and IgM Abs to recognize GP1a and GP1e in an ELISA. IgG from clone A6-9 reacted to GP1e but not GP1a (Fig. 4D). In contrast, none of the purified IgM Abs recognized either GP1a or GP1e. Taken together, these data show that the IgG from clone A6-9 can recognize the peptide sequence of the Ebola virus GP1e (mucin-like region). However, the IgM Abs from clones A13-2, C21-2, C22-2, E53-2, G3-2, G29-5, G44-2, J10-2, M65-26, N47-9, and N52-7 failed to recognize Ebola virus GP1a and G1e, suggesting that these IgM Abs were not antigen-specific. When IgG from clone A6-9 was further tested to determine its IgG subclass, it was found to be an IgG2a type (Fig. 5A). Moreover, the lowest concentration of IgG from clone A6-9 that displayed GP reactivity was determined as 1.95 ng/mL (Fig. 5B).
The production profiles of IgM- and IgG-secreting hybridomas generated after booster-injections of GP DNA vaccines or GP
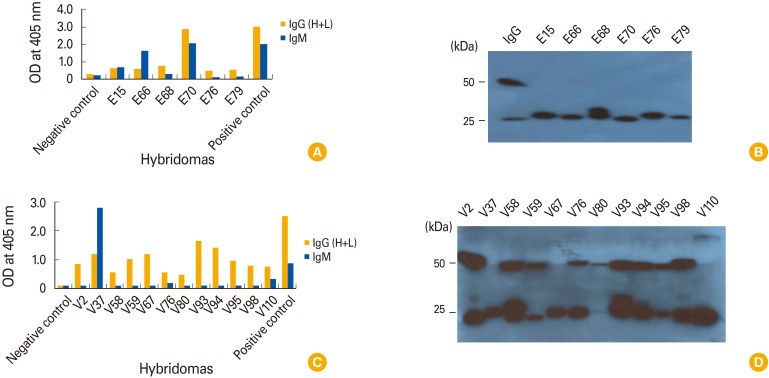
To compare the effects of the delivery method and source of GP on the production of IgM- and IgG-secreting hybridomas, we booster-injected the mice with GP DNA vaccines (by IM-EP) or GP (by intravenous delivery). GP was intravenously delivered because it was a soluble protein. For final boosting, intravenous delivery is the recommended method for soluble protein antigens [12]. After booster-injecting the mice with the GP DNA vaccines, six hybridomas (E15, E66, E68, E70, E76, and E79) were selected by ELISA (Fig. 6A). All of the selected hybridomas secreted an IgM isotype, as measured by ELISA using HRP–anti-IgG (H+L) and HRP-anti-IgM as the secondary Abs (Fig. 6A). When cell supernatants from the six hybridomas were tested for the presence of IgM by Western blot assay using HRP-conjugated anti-IgG (H+L), they all displayed only 25-kDa L chains (Fig. 6B), confirming that the six hybridomas produced IgM isotype Abs. In contrast, control IgG from clone A6-9 displayed both 50-kDa H and 25-kDa L chains. These data corroborate the notion that DNA vaccination leads to preferential production of IgM-secreting hybridomas. When the mice received booster injections of GP, they produced 12 hybridomas (V2, V37, V58, V59, V67, V76, V80, V93, V94, V95, V98, and V110) (Fig. 6C). In particular, three hybridomas (V37, V76, and V110) secreted an IgM isotype, whereas nine hybridomas (V2, V58, V59 V76, V80, V93, V94, V95, and V98) secreted IgG isotype Abs. This was determined by an ELISA using HRP-conjugated anti-IgG (H+L) and HRP-conjugated IgM as the secondary Abs. This was further confirmed by a Western blot assay (Fig. 6D). Cell supernatants from the hybridomas V2, V58, V59, V76, V80, V93, V94, V95, and V98 displayed both 50-kDa H and 25-kDa L chains, whereas those from the hybridomas V37, V67, and V110 displayed only 25-kDa L chains. These results suggest that nine hybridomas (V2, V58, V59 V76, V80, V93, V94, V95, and V98) produce IgG types, whereas two hybridomas (V37 and V110) produce an IgM type. However, the cell supernatant from the remaining hybridoma (V67) exhibited neither IgM-specific reactivity to GP (by ELISA) nor 50 kDa H chains (by Western blot assay), suggesting that hybridoma V67 might produce Abs other than IgG or IgM. In the case of the hybridoma V76, its cell supernatants exhibited both IgM-specific reactivity to GP (by ELISA) and 50-kDa H chains (by Western blot assay), suggesting that the hybridoma V76 might be polyclonal and produce both IgG and IgM. Taken together, these collective data show that protein boosting following DNA vaccination might reverse the hybridoma production profile from IgM- to IgG-secreting hybridomas.
Discussion
In this study, we observed that IgM-secreting hybridomas were preferentially produced by immunization with DNA vaccines encoding Ebola virus GP. Cell supernatants from these IgM-specific hybridoma clones displayed binding activity to different parts of GP in an ELISA, suggesting that IgM-specific hybridoma cells might generate IgM Abs with little antigenic specificity. Despite this, however, all purified IgM Abs were unable to recognize GP. Presently, it is unclear why, unlike the hybridoma cell supernatants, the purified IgM Abs exhibited no binding to GP. This phenomenon was also observed when animals were immunized with DNA vaccines coding for the surface antigens of other infectious viruses, including Middle East respiratory syndrome-causing coronaviruses and severe fever with thrombocytopenia syndrome viruses (data not included). On the other hand, the purified monoclonal IgG from one hybridoma clone was an IgG2a type with binding activity to the mucin-like region of GP. In addition, preferential production of IgM-secreting hybridomas was observed when animals were booster-injected in the foot pad with GP DNA vaccines or intravenously with tumor cells expressing GP (data not shown). This was unexpected, as we observed in this study that Abs were produced in the serum of animals immunized with GP DNA vaccines in the order of IgG2a, IgG2b, IgG1, IgM, and IgG3 in their quantity (data not included). This result implies that DNA vaccines are able to induce the production of IgG Abs in animals through a switch from IgM to IgG by B cells. In this context, it is likely that the preferential production of IgM-secreting hybridomas over IgG-secreting hybridomas by DNA vaccination might not be ascribed to a lack of B cells that switch to IgG isotypes. Instead, the nature of DNA vaccines as nonspecific immune inducers might be attributed to the production of hybridomas secreting IgM Abs with little antigen specificity. This idea is supported by the previous finding that the un-methylated CpG motifs of bacteria-derived DNA are responsible for non-specific immune stimulation and polyclonal B cell activation [1314].
We also observed that the use of GP to boost following DNA vaccine delivery led to preferential production of IgG-secreting hybridomas over IgM-secreting hybridomas. In our observation, nearly 100% of hybridomas generated by boosting with GP DNA vaccines produced an IgM type, whereas 17% of hybridomas (2/12) generated by boosting with GP produced an IgM type. Furthermore, of the 12 hybridomas obtained by boosting with GP, nine produced IgG isotype Abs in their cell supernatants, suggesting an important role of protein boosting in the preferential production of IgG-secreting hybridomas. These cell supernatants contained mostly IgG2a and, to a lesser degree, IgG2b isotypes (data not shown), implying that IgG responses might be primed by DNA vaccination. Previously, we reported that, contrary to DNA vaccines, protein vaccines were able to induce little antigen-specific IgG2a production [1516]. This finding, along with our previous findings, suggests that antigen-specific Ab responses, primed by DNA vaccination, may be boosted by protein immunization, leading to preferential production of IgG-secreting hybridomas. This notion is supported by the fact that B cells, which were used as a target for fusion for hybridoma production 3 days after protein booster injection, were unlikely to have experienced a class switch from IgM to IgG, which was needed for the production of IgG-secreting hybridomas. Indeed, we observed that no significant level of antigen-specific IgM and IgG were detectable in sera obtained at 3 days after injection with recombinant GP in the absence of priming with DNA vaccines (data not shown). This idea is also supported by the previous finding that protein priming-protein boosting had no effect on IgG2a isotype induction, whereas DNA priming-protein boosting dramatically increased production of IgG2a [15]. Thus, recombinant proteins, as a final booster vaccine, may play a role in the increased production of IgG-secreting hybridomas in the DNA vaccination platform. Taken together, these data suggest that protein boosting following DNA vaccination might be useful for generating IgG-secreting hybridomas.
In the present study, we also observed that purified monoclonal IgG Abs resolved into two protein bands (50-kDa H chains and 25-kDa L chains) on an SDS-polyacrylamide gel, but that purified monoclonal IgM Abs had differently sized bands (75-kDa H chains and 25-kDa L chains). In addition, both the H and L chains of IgG, but only the L chain of IgM were recognized by HRP-conjugated anti-IgG (H+L) in our Western blot assay. This is consistent with our ELISA data showing that IgG and IgM were both recognized by HRP-conjugated anti-IgG (H+L), but that only IgM was recognized by HRP-conjugated anti-IgM. Thus, this result shows that HRP-conjugated anti-IgG (H+L) can be utilized as a secondary Ab to measure the levels of IgM plus IgG isotypes.
GP is responsible for binding to host cell receptors for viral entry [1718]. We observed that purified monoclonal IgG from clone A6-9 recognized the mucin-like region of GP. It would also be interesting to test which parts of GP are recognized by the IgG Abs purified from our nine IgG-secreting hybridomas. Some of these Abs, in particular the IgG Abs with GP binding activity, could act as neutralizing Abs by blocking the host-virus interaction, and be further utilized to treat patients with Ebola virus infection. The reactivity of the additional IgG Abs should be tested in in vitro and animal studies.
In summary, DNA vaccination preferentially produced IgM-secreting hybridomas over IgG-secreting hybridomas. However, boosting with recombinant proteins following DNA immunization facilitated the production of a higher proportion of IgG-secreting hybridomas. Thus, this protein boosting approach may have implications for the production of IgG-specific hybridomas in the DNA vaccination platform.
 XML Download
XML Download