

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Streptococcus pneumoniae (pneumococcus) is one of the most virulent bacterial pathogens, causing many variable diseases such as pneumonia, bacteremia, and meningitis, and has high morbidity and mortality [1,2]. Though it often colonizes the nasopharnyx, pneumococcus can spread from this location to the lung, bloodstream, central nervous system, and other sites [3].
Thusfar, more than 95 capsular types of pneumococci have been described [4], and a 23-valent polysaccharide vaccine and conjugate protein vaccines with 9, 10, or 13-valent capsular polysaccharides have been developed and are currently available. However, the 23-valent polysaccharide vaccine does not elicit a memory response, and is therefore limited to use in adults, as it is not immunogenic in infants [5]. Although highly effective, conjugate vaccines only provide protection against the capsular types/groups present in the vaccine [6-8].
Several pneumococcal protein and polysaccharide virulence factors are now known to elicit protective mucosal immunity [9,10]. Specifically, most pathogens able to infect via the host's mucosal membranes evoke a local mucosal immune response, which is typically mediated by secretory IgA [11]. In a murine model, whole inactivated pneumococci were proved to be immunogenic when inoculated via mucosal membranes, thereby suggesting that the nasal mucosa may represent the preferred site for antigen delivery [12]. Moreover, nasal immunization with inactivated pneumococci may also be able to induce immunity against pathogenic pneumococci [13]. However, to date most currently-available mucosal vaccines are administered parenterally and include additional adjuvants such as cholera toxin, nontoxic subunit B, or interleukin-12 [9]. As such, a new type of pneumococcal vaccine is needed that can induce mucosal immunity and also protect against initial host colonization as well as subsequent invasive sequelae. This ideal vaccine would furthermore protect against pneumococcal invasion regardless of capsular type, and thus provide herd immunity in the population as a whole [10,14,15].
Previously, we reported that the pep27 gene plays a crucial role in pneumococcal virulence, and that pneumococci with the pep27 mutation could be used as an attenuated live vaccine [16]. However, the pep27 mutant contains an erythromycin resistance gene, which could be transferred to other commensal organisms in the nasopharynx during immunization. Therefore, in this study, the erythromycin resistance gene was deleted from pep27 mutants. As a safe and effective vaccine is required prior to the initiation of human clinical trials, this study assessed whether inoculation with the inactivated pep27 mutant could protect from a lethal pneumococcal challenge. Our results demonstrated that the inactivated pep27 mutant could protect against alethal challenge and that the pep27 mutant antisera cross-reacted with a number of heterologous strains, suggesting that the pep27 mutant is highly feasible as a mucosal vaccine.
Materials and Methods
Bacterial strains and cell culture conditions
The bacterial strains used in this study are referenced in Table 1. S. pneumonia encapsulated highly virulent capsular serotype 2 (D39) (NCTC7466; [17]), mutants of D39, serotype R6 (R6) [18], serotype 3 (WU2) [19], serotype 4 (TIGR4) [20], serotype 6B (BG7322) [21], and serotype 19F (EF3030) [22] were cultured in Todd Hewitt broth at 37℃ with 0.5% yeast extract (THY; Difco Laboratories, Detroit, MI, USA).
Construction of markerless pep27 deletion mutant
A Cheshire cassette (Genbank accession No. FJ981645) carrying the erythromycin-resistance marker (ermAM), which can be used as a temporary marker for selection [23], was amplified by primers (5'-TGG CTT ACC GTT CGT ATA G-3' and 5'-TCG ATA CCG TTC GTA TAA TGT-3'), which was received from Dr. Donald Morrison (University of Illinois at Chicago), and ligated by polymerase chain reaction (PCR) with upstream and downstream sequences amplified by primers (5'-TCT CTA TCG GCC TCA AGC AG-3' and 5'-CTA TAC GAA CGG TAA GCC A GAT TTT CAC CAC TGC TTT CG-3', and 5'-ACA TTA TAC GAA CGG TAT CGA AAG GCC AGC AAG AGA CTA-3' and 5'-CTG CGA GGC TTG CAC TGT AG-3' from genomic DNA of D39, respectively).
The ligated product was then transformed into D39 to create pep27 mutant. Competence was controlled by the appropriate addition of competence-specific peptide [24] and transformed as described previously [25]. The transformation medium was prepared from BHI broth by adding 1 mM of CaCl2. The culture was incubated at 37℃ until the O.D550 reached about 0.085-0.1. One milliliter of culture was then aseptically transferred to a sterile 1.5-mL tube, after which competence-stimulating peptide 1 was added to each tube at a final concentration of 100 ng/mL. Each tube was then incubated at 37℃ for 15 minutes in a 5%-CO2 incubator before the PCR product was added, and the reaction was gently mixed by pipetting. The tubes were then reincubated at 37℃ for 4 hours in a 5%-CO2 incubator. To select the pneumococcal transformants, erythromycin was added to the growth medium at a concentration of 0.1 µg/mL.
Cheshire cassette excision was induced by adding 1% L-fucose (Sigma, St. Louis, MO, USA). The fucose-treated cultures were then plated on THY blood agar plates to obtain single colonies. The presence of the Cheshire cassettes in each colony was confirmed by PCR using the following primers: 5'-TCT CTA TCG GCC TCA AGC AG-3' and 5'-CTG CGA GGC TTG CAC TGT AG-3'. The mutant (THpep27) sequence was confirmed by nucleotide sequencing (Cosmo, Seoul, Korea) as well as immunoblot analysis with Pep27 antibody (data not shown).
To confirm the THpep27 mutant at the RNA level, RNA from bacteria in the early exponential phase was isolated using the hot phenol method described previously [26]. After DNA was removed by DNase I (Takara, Tokyo, Japan), one microgram of bacterial RNA was reverse transcribed into cDNA using a random primer (Takara). Reverse transcription PCR was performed according to the manufacturer's instructions (Super Bio, American Building Restoration Products Inc., Franklin, WI, USA) using the recommended primers.
In vivo infection studies
Four-week-old male CD1 mice (Orient, Seongnam, Korea) were used for all infection experiments. The use of animals in the study was approved by the Sungkyunkwan University Animal Ethical Committee in accordance with the guidelines of the Korean Animal Protection Law.
Virulence studies in vivo
Intraperitoneal (i.p.) or intranasal (i.n.) challenge with D39 and its isogenic mutant was performed to evaluate the effect of the pep27 mutation on the virulence of S. pneumoniae. Bacteria were cultured overnight at 37℃ on blood agar then transferred to a serum broth (10% [vol/vol] horse serum in brain heart infusion broth [Difco Laboratories] or Todd-Hewitt broth [Difco Laboratories]) and incubated at 37℃ for 3 hours to reach a ca. 108 colony-forming unit (CFU)/mL [27]. After each bacterial culture was diluted to an appropriate density in the serum broth, groups of CD1 mice were infected with either D39 or the mutant. The quantity of broth used varied by the route of infection, with 0.1 mL used for i.p. and 10 µL for i.n. The survival of the challenged mice was then assessed four times daily for the first 5 days, twice daily for the following 5 days, and daily until 21 days post-challenge. Virulence was determined by the number of CFU required to kill 50% (LD50) of the exposed mice (Table 2).
Mucosal immunization
For immunization, mice were vaccinated i.n. (approximately 1×107-1×108) with the inactivated pep27 mutant every 2 weeks for a total of three times. One week after the third immunization, serum samples were obtained by retro-ocular bleeding and stored at -20℃ prior to enzyme-linked immunosorbent assay (ELISA) analysis. Antibody titration was performed as described previously [28].
At 2 weeks after the final immunization, the mice were challenged i.n. with 10 µL of the highly virulent S. pneumoniae D39 strain at ca. 2×107 CFU/mouse. Survival was monitored every 4 hours for 7 days, then daily for up to 21 days. Additionally, the total survival time for each specific mouse was also tracked.
Statistical analysis
All statistical analyses of antibody levels were performed using either a paired or unpaired Student's t-test. Comparisons of colonization density between groups were assessed by the Newman-Keuls multiple comparison test. Differences in survival times between groups were analyzed using the log-rank (Mantel-Cox) test. All ELISA data are expressed as the average of triplicate wells±SEM. In all cases, statistically significant differences were defined as p<0.05.
Results
Construction of a markerless pep27 mutant
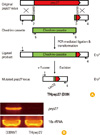
The EHpep27 pep27 mutant was previously shown to have a significantly attenuated virulence, so that immunization with this mutant effectively provided protection from lethal challenge [16]. However, as the EHpep27 pep27 mutant contains an erythromycin-resistance (Ermr) marker, it is not suitable for human clinical trials, since the Ermr marker can be transferred to commensal mucosal microorganisms. Therefore, a new pep27 mutant was created using the Cheshire cassette, which contains a temporary erythromycin-resistance marker to allow for the selection of the desired recombinant. This cassette was then subsequently removed using the embedded fucose-regulated Cre recombinase [23]. The resulting new pep27 mutant, THpep27, thus did not carry any drug resistance markers (Fig. 1A). The deletion of the pep27 gene was confirmed by nucleotide sequencing of the mutant chromosome (data not shown). A lack of pep27 gene expression was then further corroborated by analysis of mRNA expression (Fig. 1B).
Broad cross-reactivity of the pep27 mutant antisera with other pneumococcal strains
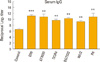
Previously, immunoglobulin titers of EHpep27 mutant anti-sera were shown to be significantly higher than non-immune normal sera, suggesting that mice immunized with EHpep27 were cross-protected from lethal challenges from heterologous strains [16]. This cross-protection further suggests that the EHpep27 mutant antisera may cross-react with other serotype strains. To confirm this relationship, mice were i.n. immunized three times with approximately 1.5×107 of THpep27 mutant, and subsequent antisera were analyzed for IgG titers to heterologous strain lysates. Titers of the THpep27 mutant antisera to EF3030 (type 19F), TIGR4 (type 4), BG7322 (type 6B), WU2 (type 3), and R6 lysate all increased significantly when compared to the non-immunized normal controls (Fig. 2), thus indicating that THpep27 antisera is able to cross-react with other heterologous strains.
Protection from lethal challenges after immunization with the inactivated pep27 mutant
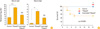
It has been previously reported that whole heat-inactivated pneumococci can stimulate both systemic and mucosal antibodies, including IgG and IgA [29], both of which represent crucial components of the host immune response to pneumococci. Therefore, to evaluate whether immunization with the inactivated THpep27 mutant is able stimulate antibodies that protect against pneumococci, mice were i.n. immunized three times with the inactivated THpep27 mutant at 2-week intervals, after which time IgG and IgA titers were quantified. Immunization with live THpep27 mutant produced significantly higher titers in comparison to non-immunized controls. Conversely, while immunization with the inactivated THpep27 mutant did elicit a rise increase in titer levels when compared to the control mice, the differences did not reach statistical significance (Fig. 3A).
When the immunized mice were challenged i.n. with lethal D39 at 2 weeks post intranasal immunization, the mice immunized either by live or inactivated THpep27 mutant were 100% protected (p=0.0088) and survival time was significantly increased (Fig. 3B). This data suggests that while immunization with either the live EHpep27 or inactivated THpep27 mutant is able to protect mice from a lethal challenge with a virulent strain, immunization with live THpep27 mutant is more effective.
Decreased colonization of D39 by the inactivated THpep27 mutant immunization
Previously, we demonstrated that the EHpep27 mutant was rapidly cleared by the host immune system and was not able to colonize the lung, blood, spleen, and brain after in vivo i.n. infection [16]. To confirm that i.n. immunization with the inactivated THpep27 mutant was also able to prevent secondary colonization with a virulent strain, mice were i.n. immunized three times with the inactivated THpep27 mutant (2×107 CFU), then subsequently i.n. challenged with approximately 1×107 of virulent D39 strain 2 weeks after the third immunization. The resulting number of viable bacteria in organs was then quantified. At three hours after infection, the virulent D39 strain colonization levels in the nasopharyx and nasal wash were 10- to 100-fold lower in the mice immunized with the inactivated THpep27 mutant when compared to the non-immunized control group (Fig. 4). Similarly, the virulent D39 strain colonization levels at 6 to 12 hours after infection were 5-fold lower in the nasopharyx and 10-fold lower in the nasal wash among the THpep27-immunized mice when compared to the non-immunized controls (Fig. 4). These results demonstrate that immunization with the inactivated THpep27 effective lyprotects against a lethal challenge in vivo.
Discussion
According to the World Health Organization, approximately 800,000 children die of invasive pneumococcal diseases each year [29]. Although currently used to reduce the morbidity and mortality of pneumococcal disease, conjugate vaccines can only protect against the specific pneumococcal serotypes included in the vaccines. While many of the causative pneumococcal serotypes responsible for invasive disease are included in the current 23-valent polysaccharide or conjugate vaccines, pneumococcal disease also affects the global health burden due to widespread multiple antibiotic resistance and serotype replacement [30]. Moreover, the relative expense of conjugate vaccines also prevents widespread immunization in developing countries, further increasing the worldwide pneumococcal burden [31]. Accordingly, inexpensive vaccines with broader serotype coverage and high immunogenicity are needed. Recently, a single i.n. immunization with a live attenuated capsular polysaccharide (cps) mutant was shown to protect against lethal challenge and provide mucosal immunity [28]. However, as this cps mutant colonized the upper respiratory tract for 9 days [28], some concern exists that this strain may have the potential to revert back to the virulent wild-type during colonization. Moreover, while inactivated unencapsulated whole cell immunization has been used i.n. as a mucosal vaccine [32], adjuvants are always required for mucosal immunity to be elicited, many of which have been associated with Bell's palsy [33]. In contrast, the pep27 mutants described here do not require any adjuvant to elicit mucosal immunity and are all cleared rapidly within 48 hours.
Although the virulence of the pep27 gene has not fully been characterized, the D39 pep27 locus in type 2 S. pneumoniae has 100% amino acid sequence homology with JJA, ATCC700 669 (Spain 23F-1), and non-encapsulated R6 strains. The pep27 family also has a homology greater than 93% with other S. pneumoniae strains, including Hungary 19A-6, TIGR4 (type 4; SP0602), and CGSP14 (type 14; SPCG0563), indicating a high prevalence of the pep27 gene among pneumococci. However, no work has been done to evaluate the effect of pep27 on virulence. The results presented here demonstrate that immunization with both live attenuated [28] and inactivated pep27 mutants protects against the nasopharyngeal colonization of lethal pneumococcal strain in mice.
Intranasal immunization with the cps mutant increased IgG antibody titers and was able provide protection against lethal challenge [34]. Our results demonstrate that i.n. immunization with live pep27 mutant also increases IgG titers and protects vaccinated mice from lethal challenge. Therefore, the pep27 mutant might behave in a way similar to that of the cps mutant in antibody production and protection from lethal challenge.
While immunization with live attenuated cps mutant is able to provide protection from a secondary lethal challenge without requiring an adjuvant [28], this strain was found to persist in the host 9 days after infection. Conversely, the pep27 mutant was rapidly cleared from the nasopharynx. That the pep27 mutant is rapidly cleared and still able to elicit mucosal immunity could result from the propensity of this strain to aggregate, thus decreasing mucosal survival rates [16]. Similarly, mice immunized with inactivated THpep27 mutant had lower nasopharyngeal colonization rates of virulent D39 when compared to non-immune controls. Since IgG and IgA titers in the mice immunized with inactivated THpep27 mutant were not significantly increased when compared with the normal control group, however, the protection conferred may result from to be due to cellular rather than humoral immunity. Clearly, further studies are warranted to clarify the exact mechanisms by which the pep27 mutant triggers and/or stimulates the immune system and thus provides protection.
In summary, pep27 mutant antisera seem to cross-react with other serotypes, and immunization with the pep27 mutant effectively eradicates colonization of virulent strains in the nasopharynx. In this way, the pep27 mutant may prove useful in developing an inexpensive attenuated vaccine able to elicit mucosal immunity.



 XML Download
XML Download