

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The influenza virus belongs to the family Orthomixoviridae and is categorized into three subclasses (A, B, and C) according to antigenic differences of two internal proteins, nucleoprotein (NP) and matrix protein (M)1. Influenza A virus infects a wide range of animals including birds and mammals, whereas type B and C viruses predominantly affect humans [1]. Influenza A viruses are further subdivided based on the antigenic features of the surface proteins, hemagglutinin (HA) and neuraminidase (NA) [1]. To date, 16 types of HA and 9 types of NA have been identified among influenza A viruses [2]. All subtypes are found in aquatic birds, suggesting that aquatic birds serve as natural reservoirs of influenza A viruses [3,4]. The influenza virus genome consists of eight segmented single-stranded RNAs with negative polarity and encodes 10 to 11 proteins depending on the strain, via alternative splicing and the overlapped reading frame on the viral mRNAs. In addition to its high propensity for mutation (antigenic drift), the segmented RNA genome enables the virus to exchange genetic materials with each other, resulting in substantial changes in its antigenicity (antigenic shift). This poses a great challenge to the development of ideal vaccines that antigenically match the circulating strain. Further, the reassortment event occasionally results in a sudden emergence of highly pathogenic strains to which the contemporary human population have little immunity, some of them causing widespread infection among humans with high mortality, such as the 1918 Spanish influenza [5]. In the last two decades, we have also witnessed several human infections with highly pathogenic avian influenza (HPAI) H5N1 viruses [6,7], raising serious concerns over the possible acquisition of direct transmissibility of the virus among humans via genetic exchange with other human-infecting viruses, such as seasonal influenza viruses and the 2009 pandemic H1N1 virus (pdmH1N1). Of further concern, it has been recently proven possible that lab-made HPAI H5N1 could transmit to and among mammalian hosts by guided genetic mutations, fuelling contentious debates over the safety of publishing detailed results [8,9].
Fortunately, vaccination has long served as a safe and effective prophylactic measure for preventing seasonal influenza virus infections and is also considered as the primary strategy against an influenza pandemic. Preventing infection by the influenza virus primarily depends on the presence of antibodies specific to the major surface proteins, HA and NA, inhibiting the initial receptor binding and the release of progeny virus particle, respectively. Inactivated influenza vaccines, inducing protective serum antibody responses, have been clinically used for humans for over 50 years against seasonal influenza viruses, and have also been developed as pre-pandemic vaccines. However, the inactivated vaccines usually require multiple administrations with the help of appropriate adjuvants to elicit sufficient serum antibody responses to effectively protect against the infection. The inactivated vaccines are delivered by parenteral routes, inducing relatively poor levels of mucosal immunity, which constitutes the first defense line against the entry of the influenza virus. In addition, the inactivated vaccines are not able to afford an effective cell mediated immunity (CMI), which is believed to be associated with cross-protection against antigenically distant strains and rapid recovery from illness.
Live attenuated influenza vaccines (LAIVs), on the other hand, offer broader and longer lasting protection against not only homologous strains but also against heterologous infections, albeit with less efficacy in general. Reassortant LAIV carries not only two surface antigens, the HA and NA of the wild type strain, but also six internal genes encoding internal constituents, which are less variable among influenza viruses than the surface antigens (Fig. 1). Although the precise contribution of each internal protein to the quality and quantity of immunogenicity of a LAIV, such as the strength and durability of protection, and the breadth of cross-reactivity against heterologous strains, remains largely unknown, they are likely to exert influence on the overall effect of vaccination with a LAIV. This may include the modulation of innate immune responses by pro-inflammatory mediators, the supply of potentially antigenic viral peptides other than the HA and NA, and T cell activation mediated by major histocompatibility complex (MHC) class I-bound viral epitopes.
The cold-adaptation of influenza viruses has been proven as a powerful means for attenuating the virulence of the virus and thus has long served as a reliable and practical platform for the generation of LAIVs against seasonal influenza viruses [10] and pandemic, such as the HPAI H5N1 [11,12] and 2009 pdmH1N1 [13,14]. The present review discusses detailed principles underlying the development of cold-adapted LAIV, and also outlines recent alternative attenuation strategies that became possible by the recent considerable advances in our understanding of the molecular virology of the influenza virus. Finally, given the choice of generating diverse LAIVs for effective defense to a variety of influenza strains, we will provide some useful guidelines for rational vaccine design for practical use.
Live Attenuated Influenza Vaccine
Advantages of LAIV
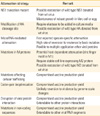
The single most important postulate of using a LAIV is that LAIV mimics natural infection and thereby presents the most effective vaccination strategy. The LAIV mounts all the phases of immune responses and thus provide the most solid protection against infection with a wild type virus. In contrast to inactivated vaccine, the LAIV is administered via the intranasal route, which is the typical entry route of influenza virus, and thus is able to stimulate mucosal immune responses, including substantial levels of secretory IgA antibodies at the respiratory tracts. Furthermore, in addition to humoral immunity mediated by antibody response specific to surface proteins, the LAIV, undergoing restricted replication in infected host, delivers a variety of intracellular viral peptides destined to be bound to MHC class I or II molecules and stimulates specific T cell repertoires. Potential advantages and disadvantages of LAIV are described in Table 1.
Classical attenuation strategies for development of LAIVs
The biggest challenge to the development of a LAIV is to render a candidate virus strain as avirulent as possible to address the safety issue of a using live virus, while maintaining a substantial level of viral growth in production hosts to elicit sufficiently high protective immune responses. Once such attenuated virus strain with desired level of safety and efficacy is successfully established, it can be reproducibly used as a donor strain for a variety of reassortant vaccines carrying suitable surface antigens, HA and NA from wild type viruses. The two major attenuation principles adopted for LAIV include the adaptation of influenza virus to nonhumans and at suboptimal temperatures, each of which is achieved through a stepwise or serial passage of the virus in either condition. The host-range vaccines generated in nonhuman cells were produced by serial passages of human influenza virus in nonhuman cells or embryonated chicken eggs [15,16]. More than 30 years ago, A/Puerto Rico/8/34 (H1N1) and A/Okuda/57 (H2N2), both of which have undergone a series of passages in nonhuman cells in the laboratory, were developed as LAIV donor strains. The reassortants based on these attenuated strains were shown to be attenuated and immunogenic in humans [16,17]. However, the reassortants in the genetic background of the A/Puerto Rico/8/34 (H1N1) showed variable degrees of virulence in some clinical trials, thus precluding their further use as LAIVs due to safety issues [18].
Avian influenza viruses as LAIV donor strains for humans also seemed to be suitable as a host-range vaccine since the avian influenza virus was genetically distinct from human strains [19,20]. Such avian-human reassortant vaccines, like the earlier host-range vaccines, showed attenuated phenotype and were immunogenic in humans [19,21], and their particular genetic constellations required for the satisfactory level of attenuation were described [22-24]. However, the avian-human reassortant vaccines also documented residual virulence, especially in seronegative infants and young children, and in some cases the generation of desired reassortant was inefficient [25], indicating genetic incompatibility between avian and human influenza viruses.
Although the two classical attenuation strategies gave way to the cold-adaptation process to establish safe, stable, and effective LAIV donor strains, they obviously provided the conceptual frameworks for subsequent advances in rational vaccine design with parallel advances in the understanding of viral pathogenesis.
Development of cold-adapted LAIV donor strains
Several animal viruses including twelve RNA viruses and two DNA viruses have been successfully adapted to grow at lower temperatures for the development of live attenuated vaccines [26]. Consequently, cold-adaptation has been considered a practical approach for the maintenance of genetic stability of live attenuated vaccines [27,28], unlike other previous strategies. Influenza viruses have also been cold-adapted by several independent groups from USA and former USSR [29-32], where A/Ann Arbor/6/60 (H2N2), B/Ann Arbor/1/66, and A/Leningrad/134/57 (H2N2) were cold-adapted by serial passages at successively lower temperatures to 25℃ in primary chicken kidney cells or embryonated chicken eggs [30,33-35]. Nucleotide sequence analysis has shown that multiple genetic alterations were introduced into all six internal genes, conferring both cold-adapted phenotype (ca) and temperature-sensitive phenotype (ts) [36-38]. It appears that stepwise or gradual lowering of incubation temperature leads to an accumulation of multiple genetic mutations in the viral genome resulting in a genetically stable variant [26, 39]. Several studies engaging reverse genetics and site-directed mutagenesis allowed significant understanding on the role of each mutation towards the attenuation phenotype of a LAIV [40-42]. Well-characterized genetic basis for attenuation and the consistent demonstration of attenuation and efficacy in diverse reassortant vaccines supported the clinical use of the cold-adapted strains. After extensive evaluation, cold-adapted A/Ann Arbor/6/60 (ca A/Ann Arbor/6/60) and cold-adapted B/Ann Arbor/1/66 (ca B/Ann Arbor/1/66) reassortant viruses have been clinically used for humans as annual trivalent seasonal influenza vaccines with promising results of safety and effectiveness [10,43]. In the late 20th century, a novel cold-adapted LAIV donor strains for influenza A and B viruses were developed in South Korea [44,45], where X-31 (A/H3N2) and B/Lee/40 were cold-adapted by serial passages in embryonated chicken eggs, and the resulting attenuated strains demonstrated acceptable level of genetic stability, safety, immunogenicity, and protective efficacy in mice.
Cold-adapted LAIVs as pandemic preparedness
In addition to the annual seasonal influenza vaccines, the cold-adapted LAIV is also considered a primary prophylactic measure against influenza pandemics, such as those by HPAI H5N1 viruses or the 2009 pdmH1N1. With the HPAI H5N1 virus emerging as a new serious threat to public health, several LAIVs against the HPAI H5N1 were generated in the genetic background of ca A/Ann Arbor/6/60 as pre-pandemic vaccines and were evaluated for their safety, immunogenicity, and protective efficacy, in various animal models including mice, ferrets, and nonhuman primates [11,12,46]. While inactivated H5N1 vaccines were shown to be poorly immunogenic requiring multiple immunizations with the help of appropriate adjuvants to elicit protective antibody responses [47,48], H5N1 LAIVs not only induced strong systemic and mucosal antibody responses against homologous strains, but also induced cross-clade protective immune responses against heterologous strains, either by a single or boost immunization without the help of an adjuvant [11,12]. The cross-reactivity of H5N1 LAIV is believed to be one of the essential requirements for providing efficient protection in the event of a pandemic, especially because of a the large diversity of antigenicity in HA of HPAI H5N1 viruses found in nature [49]. From its first reported human infection with the HPAI H5N1 virus in 1997, severe morbidity and high mortality in human cases led to a common expectation that the HPAI H5N1 might cause the next pandemic. Therefore, the sudden emergence and rapid transmission of the 2009 pdmH1N1 was rather unexpected and led us to reflect on the 1918 H1N1 Spanish flu pandemic [50-52]. Fortunately, the pdmH1N1 has caused much less fatality than expected and is now in its post-pandemic period. Nevertheless, its characteristic features such as efficient human-to-human transmission and disproportionate infection rate to children and young adults call for constant vigilance [53,54]. In addition, experimental evidence that there was considerable genetic compatibility between HPAI H5N1 and pdmH1N1 and that reassortants between the two viruses demonstrated higher transmission ability in mammalian hosts highlighted the need for vaccination prior to the global circulation of each virus or reassortants between them [55-57]. LAIVs against the pdmH1N1 were also developed using several cold-adapted donor strains including ca A/Ann Arbor/6/60, ca A/Leningrad/134/57, and ca X-31, and were shown to be immunogenic and protective in animal models [13,14,58].
Cross-protection by cold-adapted LAIVs
Antigenic drift and antigenic shift refer to the ways in which an influenza virus evades host immunity by alteration its antigenicity. Antigenic drift results from amino acid mutations in the antigenic sites of HA or NA and it abrogates the neutralizing activity of antibodies directed against previous versions of the antigens. Antigenic shift, on the other hand, occurs by genetic exchange between two or more different influenza viruses and gives rise to a novel reassortant strain. These two mechanisms present major challenges to developing an ideal influenza vaccine that could confer long-lasting immunity by a single vaccine type, necessitating annual updates of HA and NA to antigenically match newly emerged variant strain. Occasionally, as exemplified by the pdmH1N1, the antigenic shift by genetic reassortment leads to the creation of an antigenically novel virus to which most of the human population have little immunity, causing a pandemic outbreak [53,59,60]. Nevertheless, the existence of relatively less variable regions in viral proteins, such as those in the polymerase complex (polymerase basic [PB] 1, PB2, and polymerase acidic [PA]), NP, M1, or even those in the HA and NA, continually raised the possibility of developing broadly-protective influenza vaccine. LAIVs appear to better meet the requirements for notion of cross-protection than inactivated vaccines. First, the LAIV itself is in the form of a whole infectious virus and is able to carry the native conformation of all the viral constituents including surface antigens as well as internal viral components, which are subsequently processed into viral epitopes by antigen presenting cells. Many of the conserved regions of influenza viral proteins were found to carry immunogenic T cell epitopes or to be recognized by newly identified antibodies, and the prospect of utilizing such epitopes for constructing universal vaccines has been investigated by extensive studies [61-66]. Second, the LAIV is delivered via nasal route and hence is capable of inducing mucosal secretory IgA antibodies, which is more cross-reactive against heterologous strains through their polymeric nature and higher avidity [67,68]. Remarkably, LAIVs against pdmH1N1 exhibited cross-reactive immunity against antigenically distant influenza strains such as the seasonal H1N1, H3N2, and even avian H5 influenza viruses, even without detectable neutralizing antibody responses in mice and ferrets [58,69,70]. In these reports, cross-protection by LAIVs was ascribed to the induction of both cross-reactive systemic and mucosal antibody responses and to CMI. However, the detailed mechanism and relevant immune correlate of cross-protection by the LAIV remain to be fully elucidated, and the feasibility of cross-immunity in immunologically naïve human has not been addressed yet.
In light of cross-protection, establishing diverse LAIV donor strains is of particular importance since different backbone strains would differentially shape immune responses both qualitatively and quantitatively. The overall shape of immune response may include the strength and durability of protective immunity against homologous strains. Moreover, the breadth of cross-reactivity against heterologous strains is likely to be affected considerably by the distinct involvement of internal viral components in CMI. Additionally, the genetic compatibility between six internal genes from donor strain and updated surface genes from wild type virus should also be taken into account, especially for increasing the production yield of a given vaccine. Although there is no published report yet that describes such differences among currently available LAIV donor strains, it is worthwhile to construct proactively a variety of donor strains to guarantee broader protection coverage.
Reverse genetics technique in LAIV
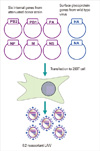
One breakthrough in the research field of influenza virus is the establishment of reverse genetics technique (RG) [71,72]. The RG allows rapid generation of a desired recombinant influenza virus from cloned cDNAs through simple DNA transfection protocols. More impressively, the RG in concert with site-directed mutagenesis made it possible to design and rescue recombinant mutant influenza viruses carrying desired mutations at any position of interest, which drove the recent advances in our understandings of molecular pathogenesis of the influenza virus. Using the RG, a reassortant LAIV can be rapidly generated by co-transfection of cDNA mixture consisting of the six internal genes of a donor strain and two surface genes from a wild type virus, obviating the need for laborious and time-consuming selection procedure used in classical co-infection followed by antibody selection of the desired 6:2 reassortant vaccine (Fig. 2). Currently, the RG system of cold-adapted LAIV donor strains has been successfully established and is used in almost all vaccine constructs including pandemic vaccines [11,73]. The technique has been recently extended to generate a novel alternative cold-adapted LAIV [74], as an alternative to the classical repeated passage at lower temperatures, allowing the conversion of a wild type virus into a genetically homologous live attenuated vaccine strain.
Alternative LAIV with modified NS1 proteins
While executing multiple accessory functions in an influenza virus-infected cell, the nonstructural protein 1 (NS1) dose not assemble into a virion particle. The NS1 protein is not obligatory to the completion of a functional infection cycle and therefore has been considered an attractive viral target for attenuating the virulence of the influenza virus and generating a novel LAIV. One of the main functions of the NS1 during the infection cycle is to play an antagonistic role against host interferon (IFN)-mediated antiviral responses, limiting both the IFN production and subsequent antiviral effects of IFN-induced proteins, such as dsRNA-dependent protein kinase R and 2',5'-oligoadenylate synthetase/RNase L [75-78]. The RG allowed the efficient rescue of a series of mutant influenza A or B viruses containing a truncated NS1 protein, which were attenuated only in IFN competent systems while maintaining growth to a high titer in cell- and egg-based substrates with defects in the type I IFN [79-81]. Thus, the NS1-mutant influenza virus emerged as a leading alternative strategy for the generation of safe and productive LAIVs. Since then, extensive studies have been conducted to characterize the biological and immunological features of the NS1 mutant viruses, and to validate their prospects for providing reliable LAIV donor stains [80,82,83]. Immunization with the NS1 mutant LAIVs were immunogenic inducing robust humoral and cell-mediated immunity in mice, pigs, horses, and macaques, and their protective immunity was not limited to homologous infection but was also cross-reactive against heterologous strains [82,84-88].
In addition to serving as a rational LAIV donor strain, the NS1 mutant virus has the potential to be a viral vaccine vector carrying multiple epitopes, whether from the influenza virus or from others, in the NS1 reading frame. Such potency of the influenza virus NS vector was experimentally demonstrated in previous studies, where the NS1 mutant influenza virus expressing the mycobacterium tuberculosis ESAT-6 protein induced CD4+ Th1 immune responses and protected animals against tuberculosis challenges [89,90]. Based on the wealth of knowledge on cross-reactive T cell epitopes of the influenza virus, it may now be possible again to generate novel universal live vaccine candidates by introduction of multiple appropriate epitopes into the NS segment.
Alternative strategies for attenuation of influenza virus
In addition to cold-adaptation and modification of NS1, several new attempts were made to attenuate the virulence of influenza virus and to generate novel live attenuated vaccine, which are summarized in Table 2.
Modification of HA cleavage site
The modification of the enzyme specificity of the HA cleavage site from trypsin-like proteases to elastase led to the generation of highly attenuated mutant strains where proteolytic processing of HA strictly depends on exogenously added elastase [91,92]. The LAIVs based on this strategy demonstrated robust immunogenicity and solid protection against lethal challenges in animal models [93-95]. While this strategy allows the conversion of any epidemic strain into a genetically homologous attenuated virus, it requires the elastase to be added to cell culture media in order to support the successful production of the vaccine virus.
MicroRNA (miRNA)-mediated attenuation
An explosive growth of scientific attention to the small RNA-mediated gene silencing mechanism was extended to vaccine technology, and serves as a toolbox for introducing stylish species-specific attenuation into an influenza virus [96]. The incorporation of nonavian miRNA response elements (MRE) into the open-reading frame of the viral NP led to an attenuation of the mutant virus in mice due to the decreased translation of viral proteins. Understandably, the viral titer in the embryonated chicken egg, which is of avian origin, was not significantly affected, obviating the need for the replacement of vaccine production host usually required to increase the production yield. However, it is possible that any secondary mutational changes in nucleotide sequences encoding the MRE can lead to reduced binding between the MRE and corresponding miRNA and consequently decrease the attenuating effect. This strategy was therefore recommended to be coupled to a pre-existing attenuating approach as a means to improve the safety level of the vaccine candidate. It is noteworthy that this strategy was the first reported attenuating method that can be applied to multiple viral targets.
Mutations in M proteins
The putative zinc finger motif in the M1 protein was reported to have little effect on viral replication in MDCK cell cultures [97], but was shown to play a critical role in virulence in mice, as the mutation of the motif caused the attenuation of the virus in mice [98]. The mutant virus that was attenuated in mice also grew poorly in cell lines derived from mice and humans, but replicated as efficiently as wild type viruses in MDCK cells and embryonated chicken eggs, suggesting host-dependent attenuation. The molecular mechanisms underlying differences in growth properties of the mutant virus among different species remain to be answered by further investigation of the biological functions of the M1 zinc finger motif.
The establishment of a stable cell line expressing the influenza viral protein made it possible to recover a replication-defective influenza virus to be used as a LAIV. Mutant influenza viruses that lacked the transmembrane and cytoplasmic tail domains of the M2 ion channel were highly restricted in growth in MDCK cells, but grew as efficiently as the wild type virus in cells stably expressing wild type M2. The virus exhibited an attenuated phenotype in mice [99], and the feasibility of the M2 knock-out mutant virus as a LAIV was examined by the reassortant pdmH1N1 vaccine candidate in a mouse model. The mutant virus elicited sterile immunity in mice completely protecting them from challenges with pdmH1N1 [100].
Mutations affecting cellular trafficking
Interfering with the cellular trafficking of influenza viral proteins during an infection became an attractive target for introducing attenuating mutations for the development of LAIVs. Mutations in the nuclear export signal of NS2/NEP or the nuclear localization signal of the M1 protein led to the attenuation of each mutant virus, immunization with each of which elicited strong immune responses and provided efficient protection against lethal challenges [101,102]. It should also be considered that the adoption of such attenuation mechanisms could lower the viral yield resulting in compromised production of a vaccine and therefore demands additional in-depth investigations on the feasibility of mass production.
Codon-pair deoptimization
Recently, genome-scale changes in codon-pair bias was presented as a novel approach to the attenuation of the poliovirus and the influenza virus [103,104]. As a result of the redundancy of their genetic code, adjacent pairs of amino acids can be encoded by as many as 36 different pairs of synonymous codons (e.g., Arginine is encoded by six different codons resulting in 6×6=36 combinations for Arg-Arg codon-pair). A species-specific codon pair bias means that some synonymous codon pairs are used more or less frequently than statistically predicted. When the codon-pairs of the poliovirus and the influenza virus were edited to contain infrequently used ones, the resulting mutant viruses were found to be drastically at tenuated by the reduced translation efficiency of mutated proteins. Since the attenuation was based on many hundreds of silent nucleotide changes across the viral genome without any alterations to its amino acid sequence, reversion to virulence is extremely unlikely and the antigenicity of the virus is not affected. Exact molecular mechanisms responsible for reduced protein translation remain to be determined. More importantly, to be a practical tool for influenza live vaccines, further investigation as to whether the attenuated virus could maintain desired level of production yield is needed.
Disruption of viral protein interaction
The disruption of the viral polymerase complex assembly by targeted mutation was tested for its potential for being a novel strategy for attenuating the influenza virus [105]. The mutations in the protein-protein binding regions of PA and PB1 disrupted the trimeric formation of the polymerase complex and decreased viral replication, finally leading to the attenuation of the virus. This strategy could possibly be extended to other interactions such as those among viral RNA, NP, and polymerase complex involved in viral replication and transcription processes, or those between the M1 and NS2/NEP complex formed during the nuclear export of viral ribonucleoprotein complexes.
Mutations in non-coding sequences
All of influenza viral RNAs carry conserved sequences at both the 5' and 3' ends and each RNA segment contains segment-specific sequences, which are partially complementary to each other to form an extended panhandle conformation [106-109]. As expected, alternative base paring in the duplex region of the conserved influenza viral RNA promoter caused attenuation of the virus [110,111]. When the G-C base pair in the duplex region of the NA segment was replaced with the U-A base pair, the mRNA and protein levels of the NA was markedly reduced, attenuating the viral replication in cells [111]. Remarkably, the attenuating effect of this base pair replacement was not specific to the NA and yet was extended to other segments tested, such as NS and PA [110], suggesting that the introduction of alternative base pairs into any of the eight segments could be used for attenuating the virulence of the influenza virus.
Suggestions for rational design of a LAIV
General considerations for rational design of a LAIV
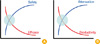
As the safety level of a live vaccine increases, the efficacy of the vaccine is likely to decrease due to the inefficient replication of the vaccine in a host. Moreover, the attenuation of viral virulence often accompanies the decrease of vaccine productivity because of the simultaneous loss of viral viability in production hosts (Fig. 3). That is, a safer and more attenuated live vaccine is likely to be less productive and efficacious. Nevertheless, as exemplified by the cold-adaptation method, it remains possible to construct an acceptably safe and attenuated LAIV with sufficient levels of efficacy and productivity. A rational vaccine design therefore should take into account these aspects simultaneously for the vaccine to be potentially practical for clinical use. For instance, establishing diverse cold-adapted donor strains could contribute to the increase of productivity or efficacy of a LAIV, whereas the introduction of a novel attenuation strategy that can be coupled to the cold adaptation may improve the safety level of the vaccine.
Directed vaccine development
Although the phenotypic contribution of each mutation accrued
during cold-adaptation of the influenza virus has been extensively investigated [40,41], the role of each mutation at the molecular level has not been fully elucidated yet. There has been only one report addressing the molecular characteristics of the PA, NP, and M1 proteins of the cold-adapted donor strain, where the PA and NP of the ca B/Ann Arbor/1/66 were found to associated with defective polymerase function, whereas the M1 protein manifested reduced membrane association in nonpermissive temperatures [112]. Molecular behaviors of wild type influenza viral proteins at higher temperatures, such as 41℃ [113], may possibly mirror those of cold-adapted strain at nonpermissive temperatures. Further investigations of the molecular determinants of the attenuation of cold-adapted strain and coupling them to another attenuation strategy would provide great opportunity toward the first step for designing "directed vaccines" [114,115].
Safety issue of LAIVs
Many of the attenuation strategies outlined above entail genetic engineering techniques involving coding or non-coding changes in the viral genome. Such modifications of viral genes present a simple and facile method for the generation of a highly attenuated influenza virus. However, it is also likely that reversion to virulence occurs by back mutation or complementing mutations during replication cycles of the vaccine virus either in the production host or in vaccinees. For instance, if attenuating mutations are introduced in only fewer positions in a particular viral protein, as in the case of the zinc finger motif of M1 [97], or cellular traffic signals in NS2/NEP [101] and M1 [102], the possibility of reversion increases accordingly.
As the influenza virus genome consists of segmented RNAs, genetic exchange among different strains occurs very frequently, which in fact has been exploited in the generation of 6:2 reassortant live vaccines. Likewise, the reassortant vaccine is also vulnerable to such reassortment with wild type strains during vaccination. Therefore, attenuation achieved through a mutation present in just one viral protein, involving either a nucleotide change or a deletion of a functional domain, is associated with a high likelihood of the restoration of virulence through donation of wild type proteins or genes from co-infected wild type virus. A possible solution is to adopt multiple attenuation markers in a vaccine either by combining different attenuation strategies or by using the same kind of mutations among different RNA segments. Resultant multiple genetic lesions, like those accumulated during cold-adaptation, would increase the genetic stability and safety level of a live vaccine by decreasing the odds of reversion to virulence. The miRNA-mediated attenuation or alteration of base pair of non-coding promoter region, for instance, could be applied to any RNA gene segment, and would provide enough flexibility of coupling various attenuation markers. The combination of multiple attenuation mutations could also be applied to the cold-adapted LAIV, especially for increasing the safety of the vaccine. It is highly likely that cold-adaptation accumulates attenuating mutations in the two surface antigens in addition to six internal RNAs, and that these mutations contribute collectively to the overall attenuation effect of the cold-adapted backbone strain.
It should be remembered that when generating a reassortant vaccine with 6:2 genetic constellation, the HA and NA of the donor strain are replaced with those of the wild type virus. While the six internal genes of the donor strain were shown to confer sufficient levels of attenuation phenotype to the reassortant vaccine, the surface antigens HA and NA originating from circulating viruses are likely to increase the virulence of the vaccine. In fact, correlations between each specific mutation and attenuation phenotype have been extensively investigated [40,41] with the exception of HA and NA genes since they would be replaced with those from wild type viruses to generate reassortant vaccines. Assuming that this hypothesis is correct, attenuation mutations could be additionally introduced into the wild type HA or NA to achieve higher levels of attenuation, further allaying the safety issue of live vaccines.
Maintaining the productivity of vaccine virus
One major barrier to the rational design of a LAIV is maintaining the productivity of the virus in the production host since the attenuation of virulence often accompanies a simultaneous reduction in the viral productivity. Cold-adapted LAIV donor strains partially overcome this problem by shifting to lower temperatures during growth in the production host. Low temperature production could be adopted in a variety of vaccine production substrates including cultured cell lines and eggs. Vero cell-cultured LAIVs have recently emerged as an alternative to the egg-based production platform in response to the question of whether egg-based vaccines would continue to meet the need for influenza vaccines, especially during an influenza pandemic involving an avian influenza strain [116,117].
Alternatively, species differences between humans and production substrates could also provide useful guidelines for devising a practical attenuation method without compromising vaccine productivity. As explained above, the miRNA-mediated attenuation of the influenza virus was achieved by employing non-avian miRNAs to selectively suppress the expression of viral proteins only in the humans, but not in the eggs.
Summary and Conclusion
With cold-adaptation at the head of list, many attenuating strategies have been proposed and advocated for designing live vaccines as alternatives to inactivated influenza vaccines. The growing body of information on influenza virology together with the RG technique rendered the design of attenuated live vaccine candidates relatively easy. At the same time, however, it provoked urgent considerations for the development of suitable vaccine production substrates in addition to embryonated chicken eggs to provide enough production yields that would be economically feasible.
In general, live attenuated vaccines were highly immunogenic, and immunization usually induced substantial level of both systemic and local mucosal antibody responses, providing excellent protection against homologous challenges. However, some approaches could not be considered practical because of poor production yield, necessitating the development of new supportive production substrates. Another concern relates to the safety issue over potential reversion to virulence either by reassortment with the circulating virus or by back mutation. Combination of various attenuation markers over multiple RNA segments is expected to increase the safety net of the designed live vaccine.
Owing to the diversity of antigenicity and the hypervariable nature of influenza viruses, eliciting cross-reactive immune responses against antigenically distinct strains became increasingly important when designing a LAIV. Establishing diverse donor strains that assume a breadth of cross-reactivity may contribute to better coverage of heterologous infections. Moreover, further identification of potently immunogenic T cell or B cell epitopes embedded in influenza viral proteins and appropriate modulation of them will enable us to generate a universal vaccine, which remains an ultimate goal of influenza vaccine development.
In conclusion, cold-adapted influenza live attenuated vaccines have shown promising results of safety and effectiveness against seasonal influenza viruses and pandemic influenza. In addition to the pre-existing cold-adapted strain, alternative approaches for designing novel attenuated live vaccines with desired levels of genetic stability, efficacy, cross-protective immune responses, and safety will expand the pool of LAIVs available for clinical use.


 XML Download
XML Download