

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
In 1959, Adams et al. first described central pontine myelinolysis as a unique clinical entity in patients who suffered from alcoholism or malnutrition and developed spastic quadriplegia, pseudobulbar palsy, and varying degrees of encephalopathy or coma from acute, noninflammatory demyelination centered within the basis pontis. Symmetrically arranged lesions of similar histology were later identified in other parts of the brain as well, principally in the thalamus, globus pallidus, putamen, and lateral geniculate body and in the white matter of the cerebellum; these were identified as extrapontine myelinolysis.13) Alcoholism and chronic nutritional deficiency were initially considered to be the causes of central pontine myelinolysis and extrapontine myelinolysis. Other causes were subsequently suggested, including hyponatremia, the treatment of hyponatremia, and hypernatremia due to sodium substitution.4,12)
Here, we describe a patient with a traumatic brain injury who developed central pontine and extrapontine myelinolysis despite appropriate correction of hyponatremia.
Case Report
A 45-year-old man was transferred to the emergency room with altered mental status after he was discovered lying on the ground. On physical examination, he showed a flexion response to pain stimuli in all extremities. His pupils were isocoric and demonstrated a normal light reflex. The initial Glasgow Coma Scale revealed E1V1M3. His blood pressure, pulse, respiration rate, and body temperature were 190/100 mm Hg, 119/min, 30/min, and 35℃, respectively. On laboratory examination, white blood cell, hemoglobin, hematocrit, and platelet count were 5290/mm3, 10.6 g/dL, 28.7%, and 82000/mm3, respectively. Serum blood urea nitrogen was 53 mg/dL (normal 10-38), and creatinine was 5.7 mg/dL (normal 0.6-1.2). Serum sodium was 108 mmol/L (normal 135-145), serum potassium was 3.2 mmol/L (normal 3.5-5.3), and chloride was 66.7 mmol/L (normal 98-110). The serum osmolality was 234 mOsm/kg (normal 185-295), and the blood sugar level was 89 mg/dL. Arterial blood gas analysis showed metabolic acidosis. His family disclosed that the patient had a history of heavy alcoholism.
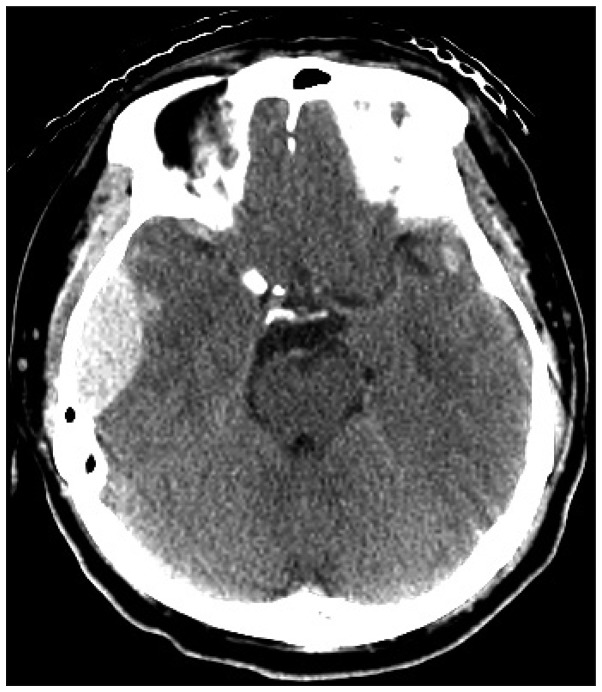
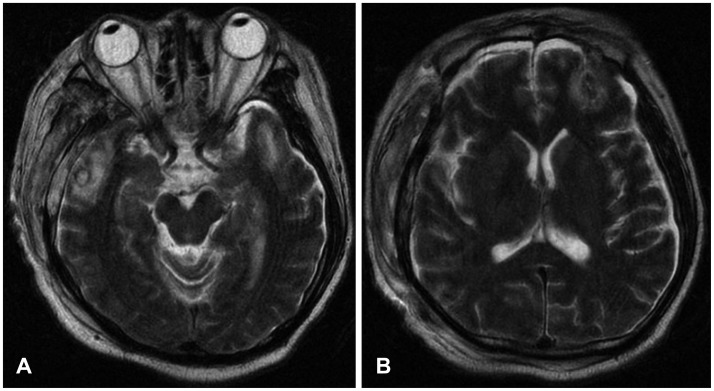
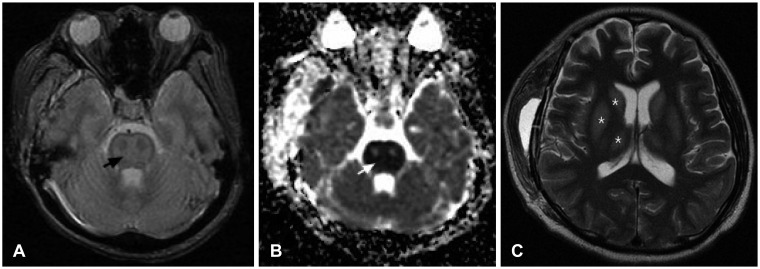
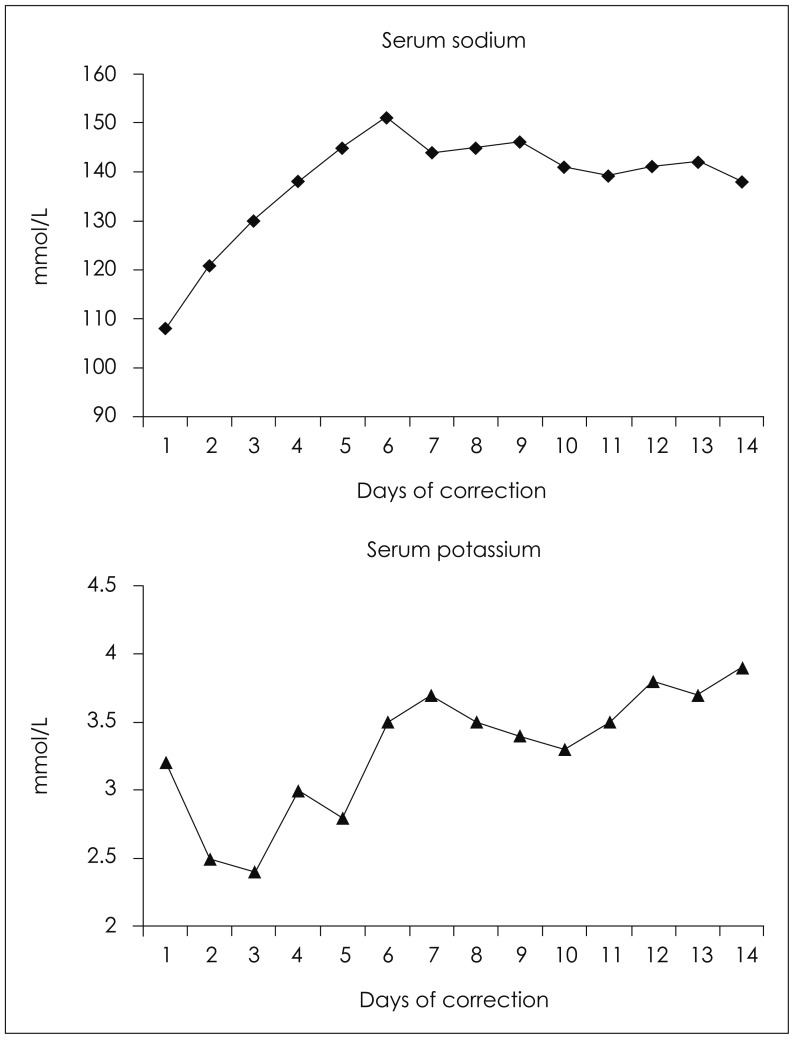
The initial brain CT scan showed a traumatic epidural hemorrhage in the right temporal region (Figure 1). As the patient was severely dehydrated and undernourished, he was started on hydration with normal saline (120 cc/hr), aiming for a sodium level of 120 mmol/L. Although his renal function recovered and his vital signs stabilized, his mental status did not improve. We performed a craniotomy to evaluate the hematoma and placed an intracranial pressure monitoring system at that time. Serum sodium and potassium were 121 and 2.5 mmol/L 24 hours after initial presentation, respectively. On the second day postoperatively, his mentation recovered to obeying to verbal commands. Serum sodium and potassium were 130 and 2.4 mmol/L, respectively. On the 4th day postoperatively, the patient was found to be stuporous. Serum sodium and potassium were 145 and 2.8 mmol/L, respectively. Brain MR images showed multiple contusions in the right temporal and left frontal regions (Figure 2). Electroencephalograghy findings were consistent with diffuse cortical dysfunction without epileptic discharge. The patient's mental status did not improve any further. On the 10th day postoperatively, diffusion weighted imagery showed high signal intensity with decreased apparent diffusion coefficient values in pons and both thalamus and frontoparietal cortex. These lesions showed high signal intensity on T2-weighted images. This finding suggested a diagnosis of pontine and extrapontine myelinolysis (Figure 3). Daily levels of serum sodium and potassium are shown in Figure 4. The patient was gradually weaned from the ventilator. His eyes spontaneously opened, but there was no improvement in cognition or lower cranial nerve palsies. He was fully dependent on a caregiver's support at the 6-month follow-up.
Discussion
Central pontine with/without extrapontine myelinolysis is a well-recognized complication of hyponatremia and rapid correction of hyponatremia. The chronicity of hyponatremia (>48 hours), sodium levels <120 mmol/L, a rapid correction of sodium (>25 mmol/L rise in 48 hours or >12 mmol/L in 24 hours), associated hypokalemia, hypoglycemia or hypoxia, malnutrition, alcohol dependency, and severe underlying illness have all been postulated to increase the risk of developing central pontine and extrapontine myelinolysis.9,10) The exact mechanism of demyelination is still unknown. One theory proposes that in regions of compact interdigitation of white and gray matter, cellular edema, which is caused by fluctuating osmotic forces, results in compression of fiber tracts and induces demyelination.12)
However, in the present patient, central pontine and extrapontine myelinolysis developed despite correction of hyponatremia was done at the speed of less than 12 mmol/L per 24 hours. Previous reports2,8) have described the occurrence of central pontine myelinolysis despite sodium correction at a rate following the above guidelines, and it has been proposed that greater caution in correction should be used (i.e., <8 mmol/L per 24 hours rise in sodium levels), especially in patients with comorbid conditions such as malnourishment due to alcoholism and chronic hyponatremia. Other factors such as the treatment of hypokalemia are also important. Myelinolysis developed in a healthy individual following an electrolyte disturbance for <72 hours.7) This may be due to concomitant hypokalemia, which was not treated before sodium correction. Reduced endothelial cell membrane concentration of NaK-ATPase in hypokalemia may predispose the cell to injury by osmotic stress associated with the rapid rise in the serum sodium concentration.3) In the present patient, hypokalemia was accompanied, but not successfully corrected.
Diagnosis is based on the detection of electrolyte abnormalities and MR findings. Because myelinolytic lesions are not demonstrated within the first 2 weeks using conventional MRI pulse sequences, the diffusion weighted image sequence has been proposed to confirm the diagnostic MRI findings of central potine myelinolysis (CPM), including symmetric signal intensity abnormalities in the central pons on T2-weighted and fluid attenuated inversion recovery imaging.11) This may progress to classic hyperintense 'trident-shaped' central pontine abnormalities, with sparing of the ventrolateral pons and corticospinal tracts. Such lesions are associated with decreased T1 signal intensity without enhancement or mass effect.6,14)
There is no specific treatment for CPM. Care is supportive with the goal of preventing complications such as aspiration pneumonia. Alcoholics are usually given vitamins to correct for other deficiencies; however, the overall prognosis is poor, and some patients die. Of the survivors, approximately one-third recover, one-third is disabled but able to live independently, and one-third is severely disabled.1,5)
Conclusion
Prevention is of utmost importance because once established, central pontine myelinolysis carries a very poor prognosis in terms of mortality and severe disability. Central pontine and extrapontine myelinolysis can occur following even gradual correction of hyponatremia, as shown in the present case. More attention should be paid to correcting hyponatremia combined with hypokalemia in patients who have a history of alcoholism.
 XML Download
XML Download