

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Granulomatosis with polyangiitis (GPA) is a life-threatening disease characterized by granulomatous inflammation and antineutrophil cytoplasmic antibody (ANCA) associated systemic vasculitis, classically involving upper and lower respiratory tracts and kidney. Milder forms of disease restricted to respiratory tract is referred to as localized GPA (loc-GPA) [12]. The European Vasculitis Study Group (EUVAS) defined loc-GPA as disease restricted to ear, nose, and throat (ENT) tract and lung without clinical signs of systemic vasculitis or disruption on vital organ function [3]. Progression from loc-GPA to systemic form is rare, occurring in only 10% of patients with loc-GPA [4]. Involvement of pituitary gland as a manifestation of progression into systemic disease in loc-GPA is also unusual, not previously reported. Here we report an unusual case of ANCA-negative loc-GPA initially presented with multiple cranial nerve palsies which progressed into ANCA-positive systemic disease involving pituitary gland.
Case
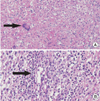
A 57-year-old woman was admitted for evaluation of polydipsia and polyuria. Two months before admission, she developed extreme thirst. She drank 3 liters of water a day to relieve her thirst. She also experienced severe generalized weakness and 3 kg of body weight loss during the 2 months period. Three years earlier, she was diagnosed with ANCA negative loc-GPA based on manifestations of recurrent pansinusitis, otomastoiditis, and multiple bilateral cranial nerve palsies (facial to hypoglossal nerve), and nasopharyngeal biopsy findings of granulomatous inflammation and vasculitis involving small-sized blood vessels (Fig. 1).
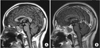
She was treated with glucocorticoid and cyclophosphamide with complete resolution of her symptoms. She was stable without recurrence for 3 years on low dose glucocorticoid and methotrexate maintenance. On physical examination, she showed severely dehydrated lips and tongue. Laboratory examinations revealed white blood cell count of 5.6×109/L, hemoglobin 14.9mg/dL, and platelet count 263×109/L. Urinalysis, and renal and hepatic functions were within normal limits. Erythrocyte sedimentation rate was normal at 20 mm/hr and C-reactive protein was negative. Hypernatremia (sodium 146 meq/L), high serum osmolarity (317 mosmol/L), and low urine osmolarity (75 mosmol/L) were found. Fluid deprivation test result suggested pituitary diabetes insipidus (DI). Hormonal levels including thyroid hormones, estrogen, luteinizing hormone, follicle-stimulating hormone, adrenocorticotropic hormone, and prolactin were within normal limits. Proteinase 3 (PR3)-ANCA was positive at a value of 31 IU/mL (normal value <3.5 IU/mL). Brain magnetic resonance imaging (MRI) was performed revealing enhancing 10.5 mm pituitary enlargement, thickened stalk, and loss of signal intensity of neurohypophysis on T1-weighted images suggestive of pituitary gland involvement of GPA (Fig. 2).
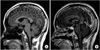
Assessment of progression from localized to systemic GPA involving pituitary gland was made. Treatment with rituximab and glucocorticoid in addition to desmopressin was initiated. Treatment resulted in resolution of polydipsia, polyuria, and thirst. Serum and urine osmolarity returned to normal values. PR3-ANCA became negative. However, follow up brain MRI in 18 months after rituximab and glucocorticoid treatment revealed persistent enhancing pituitary enlargement with minimal decrease in size (Fig. 3). She is currently under follow up on prednisolone 5 mg/day, methotrexate 15 mg weekly, and tacrolimus 1.5 mg/day with intermittent relapse in sinusitis activity.
Discussion
For purposes of conducting clinical trials, EUVAS subclassified GPA into 3 disease stages; localized, early systemic, and generalized [3]. Localized form is defined as a disease confined to respiratory tract, whereas disease extending beyond the respiratory tract is referred to as systemic/generalized form. They are further divided into early systemic and generalized form depending on presence of threatened vital organ function. Although EUVAS refers to these clinical subgroups as disease stages, it is unclear whether each subgroup is a part of disease process or represents distinct phenotype due to lack of long-term follow-up studies of loc-GPA. Only one study described long-term outcome of 50 patients with loc-GPA [4]. In this study, 10% of patients progressed into systemic/generalized disease after median 6 years supporting the stages of the disease proposed by EUVAS. Although transition rate from localized to systemic form appears to be low, this study showed that localized disease can develop into generalized disease. Low incidence of transition may suggest that effective immunosuppression at localized stage may halt the disease progression. Our patient progressed from localized disease to systemic form during the course of the disease despite treatment with glucocorticoid and cyclophosphamide suggesting that our case may represent aggressive form of loc-GPA.
There are no definite clinical features or laboratory tests that can distinguish between localized and systemic GPA. Pathologically, although biopsy specimens from localized and systemic GPA both included findings of granuloma and vasculitis, majority of the lesions in loc-GPA included granuloma, whereas systemic form included signs of vasculitis [45]. In case of our patient, pathologic findings could not distinguish between the two forms of GPA. Initial biopsy performed at localized disease stage included both granuloma and vasculitis. In addition, although biopsy could not be performed on pituitary mass, pituitary lesions in GPA patients is generally considered as granulomatous inflammation rather than vasculitis [67]. Serologically, ANCA positivity is generally considered as a clinical sign of systemic vasculitis [8]. However, approximately half of loc-GPA patients showed ANCA positivity, and ANCA positivity in loc-GPA patients was not associated with subsequent development of systemic disease [4]. In case of our patient, ANCA was initially negative, but became positive as the disease progressed into systemic form suggesting that ANCA may be an useful marker in detecting progression into systemic disease.
Pituitary involvement is a rare manifestation occurring in 1.1% of GPA patients [6]. Most common hormonal dysfunction is DI which is associated with enlargements of pituitary gland, thickening of pituitary stalk, and loss of posterior hyperintense signal on T1-weighted images on MRI [69]. Although pituitary involvement is a rare manifestation of GPA, the diagnosis was not difficult since manifestation were typical developing in a patient with existing diagnosis of loc-GPA. However, treatment was a challenge. Pituitary involvement in GPA was reported to be refractory to conventional immunosuppressive drugs with persistent hormonal and/or radiologic abnormalities despite improvement of systemic symptoms [69]. Rituximab was shown to have effect on refractory pituitary lesions of GPA [910]. We observed persistent pituitary mass even after rituximab treatment despite resolution of systemic and DI symptoms.
Our case is rare, but we believe that this case includes several important clinical implications. First is that in case of aggressive loc-GPA, it may progress into systemic disease over the disease course, thus warranting close monitoring. Second, in ANCA negative loc-GPA, conversion to ANCA positivity may reflect transition from localized to systemic disease. Finally, pituitary involvement of GPA may cause irreversible damage to pituitary gland. To minimize the risk of irreversible damage, prompt implementation of potent immunosuppressive therapy is needed.
 XML Download
XML Download