

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Pulmonary tumor thrombotic microangiopathy (PTTM) is known as a rare and severe cancer-related pulmonary complication that was first reported by Von Herbay et al. in 1990 [1]. In PTTM cases, endothelial attachment of multiple microscopic tumor emboli induces intimal proliferation of small pulmonary arteries and arterioles combined with secondary thrombosis, which leads to massive reduction of the pulmonary vascular bed and pulmonary hypertension [1,2,3]. Due to extremely rapid progression of PTTM and non-specific clinical findings, establishing a PTTM diagnosis is very difficult, and nearly all reported cases are diagnosed primarily by autopsy [1,3]. There have only been two PTTM cases reported in Korea, but they were diagnosed by autopsy or shortly before death, and thus any effective treatment was not available [4,5]. There have been only three cases worldwide that have been diagnosed ante mortem and treated effectively [6,7,8].
We report a 44-year-old woman who did not have a history of cancer but presented with rapidly progressive dyspnea and pulmonary hypertension. She was diagnosed as advanced gastric cancer and related PTTM ante mortem. She was treated effectively with chemotherapy for the underlying malignancy.
Case
In June 2013, a 44-year-old woman visited a primary clinic complaining of dry cough. She was prescribed medication for bronchitis, but the cough failed to improve. In July 2013, the symptom worsened with progressive exertional dyspnea, and she visited our hospital. She was evaluated using echocardiography and the pulmonary function test (PFT). However, there was no significant abnormality, except diastolic dysfunction (grade I: mild tricuspid regurgitation [TR]; right ventricular systolic pressure [RVSP], 27 mmHg). Her dyspnea was aggravated, and she developed left supraclavicular lymph node enlargement combined with weight loss (3 kg in a month). She was admitted to the department of hematology-oncology, KEPCO Medical Center, for further examination and laboratory tests. She had no specific medical history except for hypertension, which had been diagnosed 7 years ago. She had never smoked.
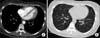
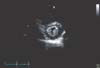
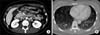
On physical examination, the patient appeared ill and had clear consciousness. Her blood pressure was 100/60 mmHg, heart rate 104 beats per min, respiratory rate 23 breaths per minute, body temperature 36.7℃, and oxygen saturation 96% (in room air). The palpated left supraclavicular lymph node was 15 mm sized, hard and firm. The results of her arterial blood gas analysis in room air were as follows: pH 7.47, pCO2 29 mmHg, pO2 91 mmHg, HCO3 21 mmol/L, SaO2 98%. Her D-dimer was elevated to 2.5 µg/mL (normal, <0.23 µg/mL), troponin I level and brain natriuretic peptide were within the normal range, 0.00 ng/mL (normal, <0.05 ng/mL) and 45 pg/mL (normal, <100 pg/mL), respectively. Tumor marker studies revealed that carcinoembryonic antigen (0.33 ng/mL) and carbohydrate antigen 19-9 (36.7 ng/mL) levels were within the normal range. A peripheral blood smear showed increased numbers of schistocytes and reticulocytes consistent with microangiopathic hemolytic anemia (MAHA) (Fig. 1). A chest radiograph showed that the lung field was clear without cardiomegaly. Electrocardiography showed a heart rate of 104 beats per min sinus tachycardia with negative T wave in V1-4, and it revealed deviation of the frontal axis to the right, and Q wave and negative T wave in III. On the 2nd day after admission, pulmonary computed tomography (CT) angiogram showed both lungs were relatively clear and no abnormalities were present in the pulmonary artery. However, enlargement of the right atrium and ventricle were observed (Fig. 2). The echocardiography showed typical findings of acute pulmonary thromboembolism with a D-shaped left ventricle, right ventricular enlargement without hypokinesia, moderate TR, and severe pulmonary hypertension with RVSP of 79 mmHg. The result was quickly aggravated by right ventricular pressure overload from the prior echocardiography (Fig. 3). Anticoagulation therapy with enoxaparin was started under the clinical diagnosis of submassive acute pulmonary thromboembolism, although there was no evidence of pulmonary thromboembolism on pulmonary CT angiography. On the 3rd day after admission, abdomino-pelvic CT and left neck lymph node biopsy were performed. Abdominopelvic CT showed a diffusely thickened gastric antrum wall and multiple enlarged lymph nodes in celiac, paraaortic, aortocaval, and mesenteric lesions (Fig. 4A). In the lung parenchyme of the abdomino-pelvic CT, ground-glass opacities and reticulonodular infiltration with tree-in-bud form had newly appeared compared with the initial pulmonary CT (Fig. 4B). On the 4th day after admission, we attempted to perform gastroduodenoscopy, which showed an ulcerofungating mass with folds converging from the low body to the proximal antrum (Fig. 5A). The pathology of the endoscopic stomach biopsy was confirmed as poorly differentiated adenocarcinoma (Fig. 5B). In addition, the lymph node biopsy showed metastatic adenocarcinoma (Fig. 5C). Therefore, gastric cancer with multiple lymph node metastases was confirmed. Although no evidence of pulmonary thromboembolism was observed on pulmonary CT angiogram and acute right ventricular pressure overload on echocardiography, we subsequently performed a lung scan. The lung ventilation scans showed no ventilation defects (Fig. 6A), while a perfusion scan showed multiple small wedge-shaped perfusion defects throughout both lungs, periphery (Fig. 6B). Therefore, PTTM caused by advanced gastric cancer was diagnosed based on clinical features, laboratory findings (including the peripheral blood smear showing MAHA), image findings (including serial echocardiography and CT images), and a lung scan. The patient's dyspnea worsened rapidly after admission, and speaking became difficult. On the 3rd day after admission, oxygen was supplied via nasal cannula. ACE-inhibitor, furosemide, and nitrate combined with enoxaparin were administered to the patient for both pulmonary hypertension and pulmonary embolism. However, her dyspnea was aggravated continuously and she demanded higher oxygen. On the 7th day after admission, 5-fluorouracil (1,000 mg/m2 continuous infusion; days 1-5) and cisplatin (60 mg/m2; day 1) were started for gastric cancer treatment. Her dyspnea was improved significantly on the 5th days after chemotherapy (12th days after admission), and we stopped her oxygen supply. She was discharged on the 8th day after chemotherapy (15th days of admission). By December 2013, she has been undergoing chemotherapy for 5 months after treatment.
Discussion
The term PTTM was first described by von Herbay et al. in 1990 as a disease entity. Earlier terms, including carcinomatous endarteritis, tumor embolization, and embolic carcinomatosis, could not fully describe the PTTM characteristics, which are hemodynamically related to fibrocellular intimal proliferation combined with secondary thrombosis [1]. The majority of the reported underlying malignancies have been adenocarcinomas of gastrointestinal origin [1,2,3,9]. Several cases with PTTM have been reported in Japan, which shows a similar incidence of gastric cancer to Korea [2,3]. However, no cases of PTTM due to gastric cancer have been reported in Korea previously. Death due to PTTM caused by gastric cancer may not be rare in Korea, and the few PTTM reports in Korea might be due to the social reluctance to undergo autopsy. In our case, gastric cancer with PTTM was identified ante mortem, and treated effectively by chemotherapy.
PTTM is characterized by tumor emboli, accelerated coagulation, and intimal proliferation [1]. Tumor emboli, in the vasculature with consequent local activation of coagulation and combined with widespread fibrocellular and fibromuscular intimal proliferation of small pulmonary arteries, leads to increased vascular resistance, resulting in marked pulmonary hypertension [1,3]. PTTM also shows hemodynamic effects similar to those observed with MAHA and disseminated intravascular coagulation (DIC). Although the underlying molecular mechanisms of PTTM remain obscure, platelet-derived growth factor, tissue factor, vascular endothelial growth factor, osteopontin and serotonin likely play roles in the pathogenesis of PTTM [2,3,6].
PTTM is difficult to diagnose, and other diseases such as infection, pulmonary thromboembolism, or drug-induced adverse events, should be excluded before diagnosis. Normal chest X-ray and chest CT findings may yield little evidence of PTTM. However, a few reports showed characteristic tree-in-bud upon chest CT [10]. In some cases, aggressive bronchoscopic biopsy, transbronchial lung biopsy, and pulmonary microvascular cytology using a wedged pulmonary artery catheter have been suggested for diagnosis of PTTM [5,6,8,11]. Due to extremely rapid progression of PTTM, nearly all reported patients have died within 1 week of dyspnea onset before differential and confirmatory diagnostic processes [2,3,9,10]. It should be noted that the aforementioned diagnostic procedures may not be applicable to patients who are already experiencing severe and progressive dyspnea. Hence, it is important for clinicians to consider the possibility of PTTM in patients with dyspnea and pulmonary hypertension based on clinical findings and non-invasive diagnostic tools in order to make an early diagnosis and provide therapeutic intervention. Peripheral blood smears for the detection of MAHA, pulmonary artery CT angiogram to exclude pulmonary embolism, and echocardiography for pulmonary hypertension are warranted. In addition, lung scans are usually not helpful given the diffuse involvement of the vasculature. However, they sometimes can show multiple peripheral subsegmental perfusion defects without ventilatory abnormalities. This finding is often referred to as the segmental contour pattern, which has been described for PTTM. In our case, although pathology was not confirmed in pulmonary vasculature, PTTM could be diagnosed based on clinical symptoms, including a stubborn dry cough and rapidly progressive dyspnea, MAHA from the peripheral blood smear, progressive changes in serial echocardiography, and peripheral perfusion defect on the lung scan. There are no specific diagnostic criteria without pathologic confirmation for PTTM due to its rarity. Considering aggressiveness of PTTM and poor tolerance of patients to invasive procedure, diagnosis using clinical symptoms and non-invasive tools might be rather appropriate in the perspective of the real practice.
PTTM is difficult to diagnose, and there is currently no effective management identified for PTTM. Chemotherapy, corticosteroids, and anticoagulation agents could be applied. Several previous studies reported PTTM cases that were determined through an ante mortem diagnosis, and the patients survived after chemotherapy [6,7,8]. Our case also showed an improvement in PTTM after appropriate management of the underlying gastric cancer. However, further studies are needed to determine whether early chemotherapy is effective, since some cases have been reported of PTTM development after chemotherapy [12].
In summary, we report a case of PTTM caused by advanced gastric cancer that improved after systemic chemotherapy. We suggest that PTTM should be considered in the differential diagnosis of patients with unexplained, rapidly-worsening dyspnea with pulmonary hypertension, even in the absence of a history of cancer.



 XML Download
XML Download