

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The most common cause of portal hypertension is liver cirrhosis and other common causes are portal vein obstruction, idiopathies or it may associate with infection, pancreatitis or abdominal trauma. Also hematological malignancies and other hypercoagulable states are rarely related with portal vein thrombosis and portal hypertension. Antiphospholipid syndrome (APS) is a clinical syndrome consists of the presence of antiphospholipid antibodies (aPL) and a status of hypercoagulability [1,2]. Intravascular thrombosis can arise in vessels of any size in any organ. APS may be divided into several categories. Primary APS occurs in patients without clinical evidence of another disease, whereas secondary APS occurs in association with autoimmune disease, infection, cancer and the use of drugs or hemodialysis [1].
We report a case of secondary APS related with systemic bacterial infection that complicated fundal variceal bleeding and ascites as a consequence of acute portal vein thrombosis.
Case
A 52-year-old man was admitted to our hospital because of chills, sweating and weight loss of 10 kg in one month. He complained of coughing and purulent sputum. He had no medical or family history of diabetes, liver diseases. He was a 20-pack-year current smoker and social drinker. He was a cook in a Chinese restaurant. At admission, his blood pressure was 130/70 mmHg, pulse rate was 108/min, body temperature was 37.3℃, respiratory rate was 22/min. His sclera was icteric and conjunctiva was anemic. Breathing sound was coarse and decreased on both lower lung fields and moist rales were heard on the left lower lung field. The abdomen was soft and slightly distended, and the liver and spleen were not palpable. The laboratory data on the admission were as follows: hemoglobin 10.1 g/dL, platelet 304,000/mm3, white blood cell count 36,300/mm3 (neutrophils 76%), ESR 120 mm/hr, CRP 18.72 mg/dL, BUN 12.3 mg/dL, creatinine 0.6 mg/dL, AST 32 IU/L, AST 17 IU/L, ALP 781 IU/dL, total protein 7.7 g/dL, albumin 1.5 g/dL, total billirubin 2.8 mg/dL, direct billirubin 2.1 mg/dL, fasting glucose 105 mg/dL, negative of venereal disease research laboratory (VDRL), negative of anti-HIV antibody, negative of HBs Ag, anti HBs and anti HCV. For evaluation of albumin:globulin (A/G) reverse, urine and serum electrophoresis (EP) was performed. There was no significant finding on urine EP. Serum EP revealed polyclonal gammopathy mixed with M-peak in gamma region, but subsequent laboratory examination showed negative serum cryoglobulin.
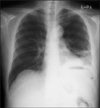
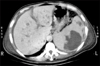
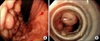
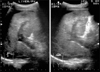
A simple X-ray showed collapsed consolidation on the left lower lung field and a air fluid level within a cavitary lesion on the left upper quadrant of the abdomen (Fig. 1). The CT revealed a large splenic abscess, multiple liver abscesses, bronchopneumonia in left lung with left pleural effusion (Fig. 2). Empiric antibiotics therapy with levofloxacin, amikacin sulfate and metronidazole was started, but the fever did not subside. Modification of antibiotics was applied to third generation cephalosporine and metronidazole. On the 8th day of admission, melena was suddenly developed. Emergency endoscopy showed isolated fundal varices in stomach with surface erosion and hematocyst (Fig. 3). Variceal bleeding was successfully controlled by endoscopic variceal band ligation. Aspirate of splenic abscess had chocolate color, foul odor and cultivated anaerobic bacteria, Eubacterium limosum. Abdomen ultrasonography revealed intrahepatic portal vein thrombosis and massive ascites with multifocal hypoechoic hepatosplenic abscesses (Fig. 4). In order to investigate the cause of the portal vein thrombosis, detailed coagulation profiles were evaluated. Anti-cardiolipin antibody IgG was 28.1 GPL (normal <20 GPL) and antineutrophil cytoplasmic antibody was positive. Prothrombin time and activated partial prothrombin time were normal, fibrinogen was 897 mg/dL (normal 170~440 mg/dL), FDP was 20 µg/mL and D-dimer was positive. Antithrombin III, lupus anticoagulant, antigenic protein C and antigenic free protein S were all negative. After bleeding stigma disappeared on second endoscopy, low molecular weight heparin (LMWH) was started for management of acute portal vein thrombosis. Since then, he had been treated with oral antibiotics for 3 months and LMWH daily subcutaneous injection for 6 months. Even after the portal vein thrombosis and abscesses were completely resolved, IgG anti-cardiolipin antibody was still elevated by 26.8 GPL. He has been regularly followed in out-patient clinic with continuous ingestion of oral anticoagulants.
Discussion
The aPL, namely, the lupus anticoagulant or anticardiolipin antibodies, are the focus of considerable interest because of their importance in the pathogenesis of clotting in the APS, a condition present not only in the autoimmune diseases, particularly systemic lupus erythematosus, but also in patients who do not manifest any overt symptoms of autoimmune disease where the emphasis is primarily on vascular events [1].
Many authors had reported raised levels of aPL in a variety infectious diseases [3-5]. Syphilis was the first infection to be linked to the aPL in 1983. And many other infections have been found to be associated with aPL positivity, but pathogenic role is not clear. HCV infection is usually not detected in patients with APS [3], but, low levels of aPL may be found in patients with HCV infection and, rarely evoke thrombotic events [4,6]. The pathogenesis of thrombotic complications in HIV infected patients and patients with AIDS is multifactorial, with the aPL playing a role in selected patients only [5,7]. Lipid disturbances consequent on antiretroviral treatment in AIDS may be prime causes of these complications. Other viral infections, including Epstein-Barr virus, varicella, cytomegalovirus, rubella virus were reported [2,8]. However, in these cases, propensity for IgM-aPL is usually shown with low titer and presence of aPL is often transient and disappears after the treatment of the infection [1,8]. Many bacterial infections demonstrate aPL. However, these increases in aPL are usually not associated with thrombotic events. Microvascular thromboses in the absence of vascular wall inflammation is a rare manifestation of leprosy, and this type of leprosy was associated with aPL [5]. aPL may also be associated with Mycoplasma pneumoniae, bacterial endocarditis and Salmonella infections were reported, but there has been some controversy about overt clinical manifestations [5].
Recently, the infectious origin of APS was proven to be one of the explanations for the generation of anti-beta-2-glycoprotien I (β2GPI) antibodies by sharing molecular mimicry with common bacteria [2,8-11]. The human β2GPI molecule, a plasma protein, has anticoagulant properties in vitro which are inhibition of prothrombinase activity, ADP-induced platelet aggregation, and platelet factor IX production [8]. The high titers of mouse anti-β2GPI were associated with an increased percentage of fetal resorptions (the equivalent of fetal loss in human APS), thrombocytopenia and prolonged activated partial thromboplastin time (aPTT), indicating the lupus anticoagulant, a presentation characterized as experimental APS [12]. Experimental APS could be induced by antibodies targeting peptides that share common epitopes with bacteria, viruses or tetanus toxoid, and the β2GPI molecule.
Most previous reports showed a high prevalence of arterial thrombosis, while portal vein thrombosis in APS has been less commonly reported and these cases were not combined infections, unlike the present case [13]. Portal vein thrombosis can occur in intra-abdominal infections, as pancreatitis, cholangitis, portal vein phlebitis and intra-abdominal abscess. In previous reports, Bacteroides bacteremia in patients with pylephlebitis suggests that the thrombogenic nature of the organism plays an integral role in the pathogenesis of septic thrombophlebitis [14]. Bacteroides species produce heparinase which may injure normal endotherial tissue in gastrointestinal organs [15]. And the lipopolysaccharide isolated from Bacteroides species accelerated coagulation in mice and activated Hageman factor [16]. These activations can play a role in promoting intravascular thrombosis. There is no proven data, in APS, that anaerobic bacteria trigger thrombosis. But there is a case report that showed portal vein thrombosis with anaerobe Bacteroides induced pyelophlebitis and transient elevation of IgM aPL [17].
Our case is unique because anaerobe induced intra-abdominal abscess complicated overt APS that IgG aPL was detected and sustained after treatment and produced portal vein thrombosis and complicated variceal bleeding and massive ascites. We used low molecular heparin instead of oral anticoagulants for resolving portal vein thrombosis in order to avoid drug interaction between warfarin and metronidazole. However, in case of secondary APS associated with infection, intensity of anticoagulation and the timing of its discontinuation are still unclear. Although the infection was controlled and portal vein thrombosis disappeared, careful monitoring will be required due to recurrence.
 XML Download
XML Download