

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pseudohypoparathyroidism (PHP) is characterized by peripheral parathyroid hormone (PTH) resistance along with hypocalce Pseudohypoparathyroidism (PHP) is characterized by peripheral parathyroid hormone (PTH) resistance along with hypocalcemia and hyperphosphatemia [1]. The identification of the PTH receptor and its signal transduction pathway facilitated better understanding of PHP pathophysiology. PHP types I and II are distinguished by cyclic AMP (cAMP) levels in response to exogenous PTH [2]. The main PHP subtypes, Ia and Ib (PHP-Ia and PHP-Ib, respectively), are caused by genetic and/or epigenetic alterations within or upstream of the GNAS locus. Most patients with PHP-Ia exhibit Albright's hereditary osteodystrophy (AHO) and multi-hormone resistance, while 2/3 of patients with PHP-Ib exhibit only PTH resistance [1]. PHP exemplifies a quite unusual form of hormone resistance as the molecular cause is not a deficiency of the hormone receptor itself, but instead is a partial deficiency of the α-subunit of the stimulatory G protein (Gsα), encoded by the GNAS imprinted locus (MIM#139320), a key regulator of the hormone-activated cAMP signaling pathway. The GNAS complex locus has multiple promoters and differentially methylated regions (DMRs) and produces several parent-of-origin products [3]. Using parent-specific methylation patterns in the DMRs, the GNAS complex produces stimulatory G protein, neuroendocrine secretory protein 55 (NESP55), the extra-large variant of Gsα (XLαs), A/B transcript, and the GNAS antisense (AS) transcript (Fig. 1) [3]. PHP-Ia is mainly caused by inactivation of maternally inherited mutations affecting Gsα coding exons. Loss of imprinting in the exon A/B DMR due to microdeletions in STX16 is the most frequent mechanism of familial PHP-Ib [45678], while the most sporadic cases of PHP-Ib are caused by imprinting abnormalities of DMRs in the GNAS complex [4910]; however, no clinical difference was observed between the familial and sporadic forms of PHP-Ib [11]. Endocrine levels and other blood biochemical parameters vary according to the stage of life and disease severity [1]. Although patients with PHP-Ia typically express AHO features, physical features, as well as biochemical and molecular findings in some cases are too ambiguous to distinguish it from PHP-Ib. The long-term effects of elevated PTH levels on the bones are also unclear [1] because various skeletal phenotypes have been observed [12131415]. Therefore, identification of the molecular cause of PHP aids in confirmation of the diagnosis and the understanding of clinical characteristics. In this report, we present a patient with PHP-Ib caused by impaired imprinting in the GNAS complex.
CASE
1. Patient
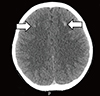
An 11-yr-old boy presented with poor oral intake and cramping pain in both the lower limbs after physical activity. The parents expressed concern that the patient had a slight physique compared to other children in the same age group. However, the patient show ed a normal growth rate according to the Korean growth chart (height =142.6 cm, 25–50th percentile, SDS −0.49; body wei ght=34.7 kg, 10–25th percentile, SDS −0.78). The patient had no family history of any relevant diseases. The patient's blood biochemical investigations revealed hypocalcemia (6.6 mg/dL, reference range 8.0–10.0 mg/dL) and hyperphosphatemia (7.6 mg/dL, reference range 2.6–4.5 mg/dL) with an elevated PTH level (127 pg/mL, reference range 13–54 pg/mL). Thyroid stimulating hormone (TSH) level was increased (8.42 mIU/L, reference range 0.17–4.05 mIU/L), but free T4 (1.14 ng/dL, reference range 0.85–1.86 ng/dL) and T3 (1.20 ng/mL, reference range 0.78–1.82 ng/mL) levels were within the normal range. Serum cortisol (5.41 µg/dL, reference range 1.81–12.67 µg/dL), 17α-OH progesterone (4 ng/dL, reference range ≤20 ng/dL), and adrenocorticotropic hormone (ACTH, 33.89 pg/mL, reference range 6.00–56.70 pg/mL) levels were also normal. To rule out PHP, further evaluations were performed. Ultrasonography and a scan (Tc-99m) of the thyroid and parathyroid glands were performed, but no abnormal findings were found. There was no evidence of AHO features in either the hand X-rays or physical examination of the patient. However, computed tomography (CT) scan of the brain revealed multiple high-density nodular lesions in the bilateral basal ganglia and subcortical white matter of the frontal lobe (Fig. 2). The cAMP level in the 24-hr urine sample was slight ly decreased (1.60 µmol/day, reference range 1.8–6.3 µmol/day).
2. Genetic Evaluation
First, we performed direct sequencing of GNAS and STX16. Amplification of exons was performed using primers designed to anneal sequences in the pre- and post-exon introns. PCR products were bidirectionally sequenced with the Big-Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) using the ABI PRISM 3130xl Genetic Analyzer (Applied Bio-systems). The Sequencher program (GeneCodes Corp., Ann Arbor, MI, USA) was used to align the derived and reference sequences (NM_000516.4). In the direct sequencing of GNAS and STX16, no pathogenic mutations were detected.
A multiplex ligation-dependent probe amplification (MLPA) assay was performed to identify the presence of deletions and duplications, including DMRs, in the GNAS complex and STX16. Methylation-specific-MLPA (MS-MLPA) was performed to identify any abnormal methylation of the sequence and copy-number changes within the GNAS complex and STX16. MLPA and MS-MLPA were performed using the SALSA MS-MLPA probemix ME031-B1 GNAS kit (MRC-Holland, Amsterdam, The Netherlands) according to the manufacturer's protocol. The ME031-B1 probemix contains 25 pro-bes specific for the GNAS locus and six probes specific for STX16 with amplicon sizes between 125 and 500 nucleotides. DNA was denatured at 98℃ for 5 min and hybridized with the probe set overnight at 60℃. The ligation reaction using ligase was performed for 30 min at 48℃, followed by 5 min at 98℃ for heat inactivation of the enzyme. PCR was performed with the specific SALSA PCR primers for 35 cycles (95℃ for 30 sec; 60℃ for 30 sec; 72℃ for 1 min). MLPA fragment analysis data was generated using the Applied Biosystems 3130xl Genetic Analyzer and analyzed using GeneMarker software (SoftGenetics, State College, PA, USA). A sample from a healthy male was used as the normal control in both MLPA and MS-MLPA assays.
The results of the MLPA analysis revealed a normal copy number for both the GNAS complex and STX16. For the MS-MLPA assay, the restriction enzyme HhaI was used to assess methylation status within a given sample. Un-methylated DNAs were digested by HhaI, and the digested probes, not amplified by PCR, did not generate a signal. In the MS-MLPA, the control sample showed a 50% reduction of each DMR (NESP55, NESPAS, XL, A/B domains), which is consistent with the previously reported normal methylation pattern (Fig. 3). In the patient's sample, the NESP55 peak was 100% preserved, but peaks of NESPAS, XL, and A/B domains show ed a 100% reduction, compared to those in the non-HhaI treated sample. This suggests gain of methylation in the NESP55 domain and loss of methylation in the NESPAS, XL, and A/B domains of the maternal allele.
DISCUSSION
PHP presents different disease features according to its subtypes owing to genetic and epigenetic aberrations in the GNAS complex [13]. The GNAS complex, a highly imprinted region, provides a better understanding of the pathogenesis of this disease. PHP-Ia and PHP-Ib are caused by distinct mechanisms, but some clinically overlapping cases have been reported [8]. In cases with an uncertain diagnosis, careful clinical examination, including assessment of AHO-specific manifestations and further laboratory and radiological investigations, are needed. Blood biochemical parameters and urinary calcium excretion should be monitored in patients with PHP-I. In case of children, height, growth velocity, and pubertal development should be closely observed. High birth weight and/or early-onset obesity and macrocephaly are features of paternal uniparental disomy (UPD) of chromosome 20q. The most sporadic cases of PHP-Ib demonstrate broad GNAS methylation defects, and no specific gene mutation has been identified as a cause of the methylation defects. Some cases of PHP-Ib have been identified to have paternal UPD as the underlying cause (10–25% according to various reports) [161819].
In Korea, several cases of PHP-Ib have been previously reported [20]. Cho et al. described six patients with PHP-Ib exhibiting various symptoms at diagnosis, such as seizures or carpal spasm. Two of the patients with PHP-Ib also showed intracranial calcifications that were revealed by a brain CT scan. Molecular studies also revealed methylation defects of the GNAS DMRs and STX16 deletions in the patients with PHP-Ib. Cho et al. suggested a diagnostic progression of direct sequencing of the GNAS gene followed by MS-MLPA of both GNAS DMRs and STX16 for the diagnosis of patients without AHO features. Microsatellite analyses were recommended to exclude paternal disomy if parental DNA was available. Garin et al. recommended single CpG bisulphite-based methods to confirm the results in patients with partial methylation defects [19]. A pyrosequencing method with bisulphite treatment is useful for the detection of more subtle methylation defects. However, MS-MLPA has the advantage of providing quantification of methylation along with detection of gene deletions and duplications. MS-MLPA analysis consists of two parts—determining copy numbers by comparing different undigested samples, and determining methylation patterns by comparing each undigested sample to its digested counterpart. The second part is unique to MS-MLPA probe mixes and serves to semi-quantify the percentage of methylation within the given sample. In our case, we performed simultaneous direct sequencing of GNAS and STX16 because the patient had no familial history and had ambiguous clinical features, such as high-density lesions revealed by a brain CT scan. After direct sequencing, MS-MLPA was performed, but without parental DNA owing to its unavailability. In previously reported Korean cases of PHP-Ib and in our case, the most frequent aberrant methylation pattern was gain of methylation in the NESP55 domain and loss of methylation in the NESPAS, XL, and A/B domains. The cases of PHP are not sufficient to prepare an appropriate diagnostic work-flow; it only allows us to ascertain the origin of the disease and provides the evidence for further genetic counseling. Each study presents a different work-flow in use for PHP diagnosis, but all studies emphasize the use of MS-MLPA. In our case, the patients showed elevated TSH levels and normal free T4 and T3 levels. TSH resistance is frequently noted in patients with PHP-Ia, while less commonly in patients with PHP-Ib. Compared to previously reported Korean cases of PHP-Ib [20], our case showed significantly low PTH levels (127 mmol/L vs. 306.3±119.1 mmol/L, P=0.014) and relatively high TSH levels (8.42 mIU/L vs. 5.75±5.71 mIU/L, P=0.3). Intracranial calcification can be observed in both subtypes. Overlapping laboratory and physical findings in PHP subtypes have made differential diagnosis challenging. Nonetheless, molecular analyses are reliable methods for PHP diagnosis. In conclusion, in addition to the GNAS and STX16 analyses using direct sequencing, quantitative analysis of gene copy number and detection of methylation defects are required for the diagnosis of PHP. Thus, MS-MLPA is a suitable method for identifying and understanding the disease.



 XML Download
XML Download