

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Joubert syndrome and Joubert syndrome-related disorders (JSRDs) are rare autosomal recessive or X-linked disorders characterized by cerebellar vermis hypoplasia and a brain stem malformation, which presents as the “molar tooth sign” in magnetic resonance imaging (MRI). JSRDs are a group of clinically heterogeneous conditions that exhibit neurological manifestations and multiple organ involvement. JSRDs are also genetically heterogeneous, and approximately 20 causative genes that account for 45% of JSRDs have been identified. A 7-yr-old boy visited Wonkwang University Sanbon Hospital with the following presentations: no ocular fixation, ataxia, growth retardation, and hypotonia. Physical examination revealed facial dysmorphism, spindle shaped fingers, and height (99 cm) and weight (13 kg) below the third percentile. Ophthalmic examination revealed retinal dystrophy. A diagnosis of JSRDs was made based on clinical and brain MRI findings. We found two heterozygous variants c.2945 G>T; p.Arg982Met (G>T) and c.2216dupA; p.Phe740Valfs∗2 (dupA) in AHI1, and a heterozygous c.3973C>T; p.Arg1325Trp (C>T) variant in KIF7 by whole exome sequencing (WES). Genetic analysis on the proband's father revealed that he had both AHI1 variants, but did not have the KIF7 variant, which was inconsistent with autosomal recessive inheritance. Therefore, the G>T variant and C>T variant were presumed to be of “uncertain significance.” Furthermore, one novel dupA variant was interpreted as “pathogenic,” while the second allele was not detected. Caution should be exercised while interpreting the significance of variants detected by WES. In addition, the involvement of genes other than the 20 known ones will require further investigation to elucidate the pathogenesis of JSRDs.
REFERENCES
1. Brancati F, Dallapiccola B, Valente EM. Joubert syndrome and related disorders. Orphanet J Rare Dis. 2010; 5:20.

2. Valente EM, Brancati F, Boltshauser E, Dallapiccola B. Clinical utility gene card for: Joubert syndrome – update 2013. Eur J Hum Genet. 2013; 21.
3. Akizu N, Silhavy JL, Rosti RO, Scott E, Fenstermaker AG, Schroth J, et al. Mutations in CSPP1 lead to classical Joubert syndrome. Am J Hum Genet. 2014; 94:80–6.
4. Tsurusaki Y, Kobayashi Y, Hisano M, Ito S, Doi H, Nakashima M, et al. The diagnostic utility of exome sequencing in Joubert syndrome and related disorders. J Hum Genet. 2013; 58:113–5.
5. Kaname T, Yanagi K, Naritomi K. A commentary on the diagnostic utility of exome sequencing in Joubert syndrome and related disorders. J Hum Genet. 2013; 58:57.
6. Chen Z, Wang JL, Tang BS, Sun ZF, Shi YT, Shen L, et al. Using next-generation sequencing as a genetic diagnostic tool in rare autosomal recessive neurologic Mendelian disorders. Neurobiol Aging. 2013; 34:e11–7.
7. Eun MY, Seok HY, Kwon DY, Park MH, So-Hee E, Kang YS. Joubert syndrome presenting with young-age onset ischemic stroke: a possible etiologic association. J Child Neurol. 2011; 26:381–4.
8. Kim JT, Kim SJ, Joo CU, Cho SC, Lee DY. Joubert syndrome with peripheral dysostosis – a case report of long term followup. Korean J Pediatr. 2007; 50:315–8.
9. Jeong HB, Hwang SH, Kim KJ, Hwang YS, Kim SC, Kim IO. Clinical study of symptoms and various anomalies of patients with Joubert syndrome. Korean J Pediatr. 1997; 40:385–92.
10. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–24.
11. Parisi MA, Doherty D, Eckert ML, Shaw DW, Ozyurek H, Aysun S, et al. AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome. J Med Genet. 2006; 43:334–9.
12. Elsayed SM, Phillips JB, Heller R, Thoenes M, Elsobky E, Nurnberg G, et al. Non-manifesting AHI1 truncations indicate localized loss of function tolerance in a severe Mendelian disease gene. Hum Mol Genet. 2015; 24:2594–603.
13. Ali BR, Silhavy JL, Akawi NA, Gleeson JG, Al-Gazali L. A mutation in KIF7 is responsible for the autosomal recessive syndrome of macrocephaly, multiple epiphyseal dysplasia and distinctive facial appearance. Orphanet J Rare Dis. 2012; 7:27.
Fig. 1.
Brain magnetic resonance imaging showing the typical “molar tooth sign” attributed to cerebellar vermis hypoplasia and brainstem malformation.
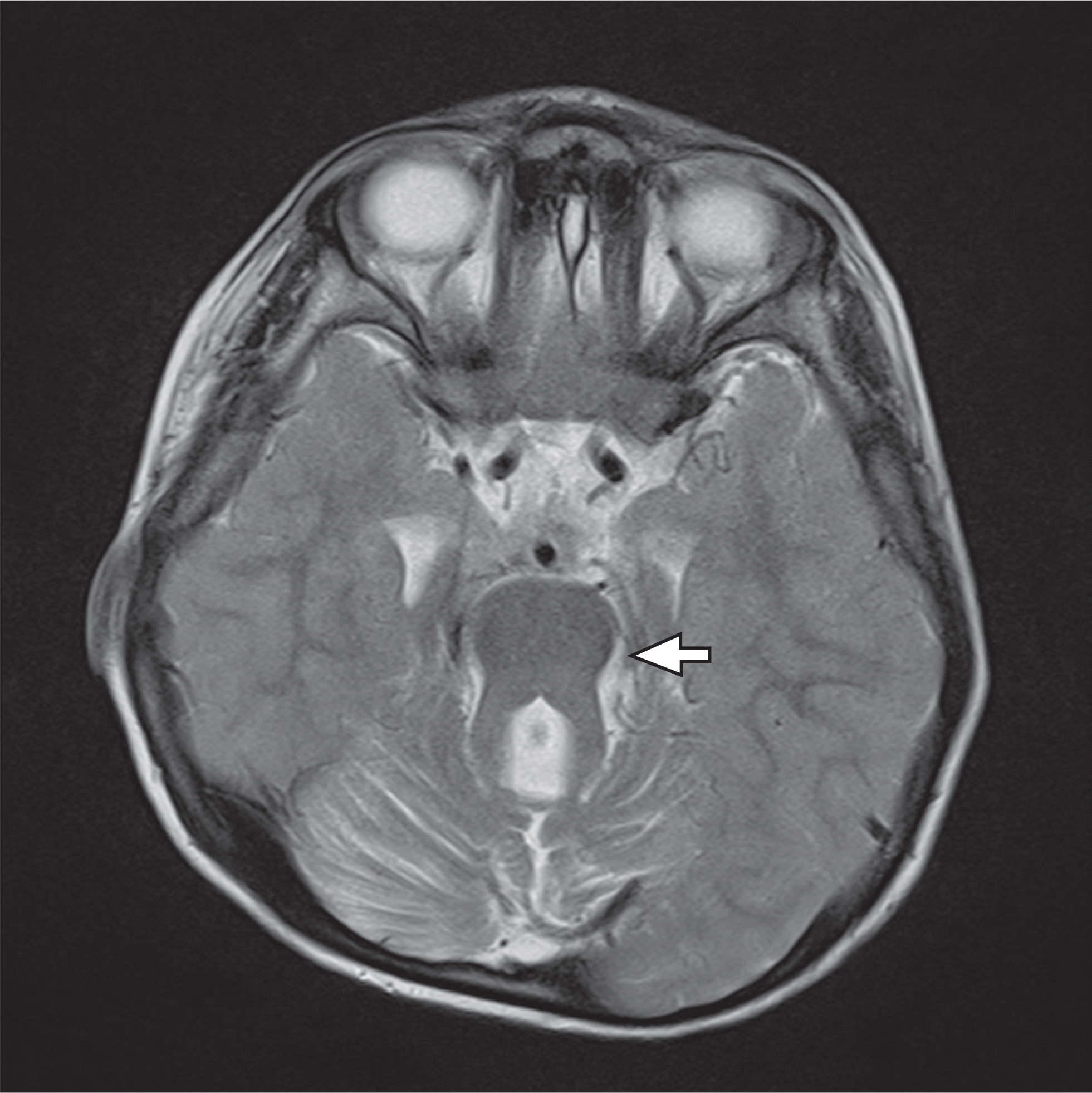
Fig. 2.
Sequencing data from the variant alleles: (A) c.3973C>T (C>T) variant of KIF7 and (B) c.2945G>T (G>T) and c.2216dupA (dupA) variants of AHI1 were confirmed by Sanger sequencing in the proband and his father.Abbreviations: w/t, wild type; NT, no test.
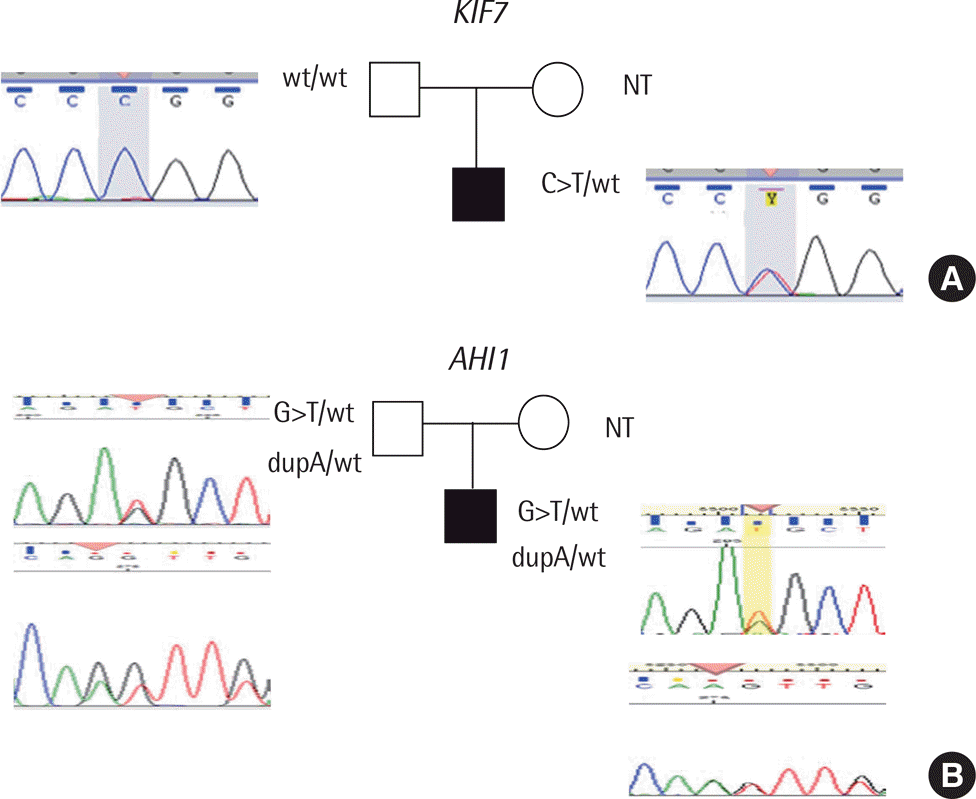
Table 1.
Joubert syndrome (JBTS)-related genes
 XML Download
XML Download