

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Neurofibromatosis type 1 (NF1) is a rare (approximately 1 per 3,500 individuals) genetic disease that transmitted by autosomal dominant pattern and fully penetrant trait.1 NF1 is caused by heterozygous mutation of NF1 gene on chromosome 17.2 NF1 has various clinical manifestation, but characterized by café au lait spots, neurofibromas, and axillary or groin freckling. Even the pathogenesis is unclear, NF 1 can cause vasculopathy.3 Vasculopathies in patients with NF1 are found in medium and large sized arteries and veins. NF1 vasculopathy usually involves cerebral, coronary, renal, peripheral vessels and causes stenosis, occlusion, aneurysmal change, rupture.2 In some cases of PH in patients with NF1, pathologic features of pulmonary artery were similar with that of NF1 vasculopathy. Therefore PH in patients with NF1 may be caused by NF1 vasculopathy.34 This kind of NF1 vasculopathy is uncommon and under-recognized complication. In the updated clinical classification of PH,5 PH with NF1 was categorized as Group 5, PH due to unclear multifactorial mechanisms. Precapillary pulmonary hypertension (PH) is an extremely severe complication and responsible for considerable morbidity and mortality.6 But the incidence of PH with NF1 is very rare, only a few cases have been reported.
We report the first Korean case of NF1 patient with PH definitely confirmed by right heart catheterization and treated with endothelin receptor antagonist.
CASE
A 65-year-old woman was admitted in our hospital with 3-year history of gradually worsening dyspnea on exertion. At initial assessment she was suffered from dyspnea which is compatible with New York Heart Association functional class III-IV. In 1993, she was diagnosed as NF 1 at other hospital. Her son was also diagnosed as NF 1. However, he did not complain of respiratory symptoms. She had undergone palliative radiation therapy for mediastinal Schwannoma in 2003 and laparoscopic right hemi-colectomy for high grade adenoma of colon in 2009. She denied the use of alcohol, tobacco or other illicit drug and the exposure of specific toxin.
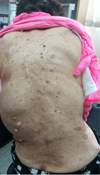
On admission, her blood pressure, pulse rate, and respiratory rate were 110/60 mmHg, 104 beats per minutes, and 22 breaths per minute, respectively. The initial physical exam revealed slightly distended both side of jugular veins and hepatomegaly. She had numerous neurofibromas on whole body especially face and trunk, which were reconfirmed by skin biopsy in our hospital (Fig. 1). She also had café-au-lait macules. Chest examination revealed grade 2/6 systolic murmur in the left low parasternal area, more accentuated P2 than A2, and crackles on both lower lung fields.
Laboratory findings on admission day demonstrated a normal complete blood count, as well as normal levels of blood urea nitrogen, creatinine, total protein, albumin, total bilirubin, aspartate transaminase, alanine transaminase, γ-glutamyl transpeptidase, and cardiac troponin I. While the patient was breathing ambient air, PaO2 and PaCO2 were 45.8 mmHg and 32.3 mmHg, respectively. The levels of anti-nuclear antibody, anti-deoxyribonucleic acid enzyme linked immunosorbent assay, P-antineutrophilic cytoplasmic antibody (ANCA), C-ANCA, anti-Scl 70, rheumatoid factor and lupus anticoagulant were negative. Anti-human immunodeficiency virus antibodies, hepatitis B virus antigen, and anti-hepatitis C antibodies were also negative.
Chest X-ray showed bilateral haziness, soft tissue mass and the scoliosis of thoracic spine (Fig. 2). High resolution computed tomography (CT) demonstrated dilated pulmonary arteries, neurogenic tumor on left parasternal area and ground-glass opacities on both lung fields (Fig. 3). Lung perfusion scan and chest CT did not show abnormal findings compatible with pulmonary embolism (Fig. 4). Pulmonary function test (PFT) revealed forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) ratio 0.80, reduced FEV1 at 75% of theoretical values, FVC at 69%, total lung capacity at 68%, and diffusion capacity of carbon monoxide at 52%. Transthoracic echocardiography showed mildly enlarged and mildly thickened right ventricle free wall, grade II tricuspid regurgitation and peak tricuspid regurgitation jet velocity 3.98 m/s with an estimated right ventricular systolic pressure 68.4 mmHg. Echocardiography excluded any other congenital, valvular and myocardial diseases.
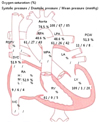
Right heart catheterization revealed pulmonary artery pressure as 61/27 mmHg (mean 43 mmHg) with mean pulmonary capillary wedge pressure of 8 mmHg (Fig. 5). Cardiac index was calculated as 2.61 L/min/m2 and pulmonary vascular resistance was 9 Wood units. Acute vasodilator test with adenosine was negative. The lowest mean pulmonary arterial pressure was 40 mmHg with maximal adenosine challenge.
Considering her clinical feature and examination findings, she could be clearly diagnosed as PH associated with NF1. She was treated with endothelin receptor antagonist (Bosentan - Actelion Pharmaceuticals, Japan) 62.5 mg twice a day for initial one month and 125 mg twice a day for the following months, diuretics and home oxygen therapy. Follow-up transthoracic echocardiography after 9 month treatment showed no significant interval change of peak tricuspid regurgitation jet velocity 3.88 m/s with an estimated right ventricular systolic pressure 65.2 mmHg. Equally her dyspnea was not significantly improved and she is being followed up uneventfully at outpatient clinic.
DISCUSSION
Our case met the National Institutes of Health diagnostic criteria for NF1 with typical clinical presentation including at least several neurofibromatosis and café au lait spots.7 Montani et al described clinical features of PH in 8 patients with NF1.8 PH in patients with NF1 was characterized by a late onset, female predominance, severe functional and hemodynamic impairment, and poor outcome. These clinical characteristics were in accordance with our case.
Someone can hypothesize that precapillary PH observed in our NF1 patient might have been caused due to parenchymal lung disease based on mild restrictive pattern revealed by PFT and high resolution CT findings. Because it has been demonstrated that NF1 may be associated with parenchymal lung involvement.9 Pathologic assessment of explanted lungs in NF1 patients who underwent lung transplantation demonstrated interstitial fibrosis with partial loss of parenchymal architecture as well as pronounced pulmonary arterial remodeling.8 But our patient complained of disproportionately severe dyspnea when considering only mild restrictive pattern on PFT. Diffusion capacity of carbon monoxide was markedly decreased suggesting significant pulmonary vascular involvement. Even her previous PFT result measured in 2011 showed mild restrictive pattern, arterial hypoxia was not observed. Thus mild restrictive pattern of PFT was not related to hypoxemia. Scoliosis may contribute to the PFT abnormality but it was not so severe as to cause hypoxic pulmonary vasoconstriction.
There is no specific treatment for PH in patients with NF1. The impact of conventional (diuretics, anticoagulation, oxygen if needed) and specific therapy including endothelin receptor antagonists, phosphodiesterase type 5 inhibitors, and prostanoids is limited.38 The limited response to PH therapies and the poor prognosis of NF1 associated PH warrant the importance of more aggressive treatment such as lung transplantation early in the course of disease.



 XML Download
XML Download