

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Kearns-Sayre syndrome (KSS) is a rare multisystem mitochondrial disorder associated with the phenotypic triad of progressive external ophthalmoplegia, atypical pigmentary degeneration of the retina, and complete heart block. Other features include sensorineural deafness, impaired intellectual function, short stature, and endocrine and renal abnormalities, which usually present before the age of 20.123 Diagnostic tools have not been definitively established, but physicians have diagnosed KSS based on many modalities including laboratory findings, electrocardiogram (ECG),4 clinical manifestations, and muscle biopsy.5
In this report, we describe a patient with type I diabetes and KSS who experienced syncope in the presence of complete atrioventricular (AV) block.
CASE
A 16-year-old boy was first seen in the emergency department with an episode of loss of consciousness. His parents reported that it was the first such episode, no premonitory symptoms had been observed, and there was no notable family history.
Physical examination revealed no abnormality and stable vital signs (blood pressure: 110/70 mmHg, respiratory rate: 18 breaths per minute, body temperature: 36.2℃), but the patient's ECG revealed complete AV block with a wide QRS (150 ms), ventricular rhythm was 34 beats per minute, and corrected QT interval was 378 ms (Fig. 1A) Therefore, we performed temporary pacemaker insertion. We observed patients in the intensive care unit (ICU) under continuous ECG monitoring without Holter monitoring.
The patient had been of short stature since childhood and did not like to exercise because of easy fatigue. At 4 years old, exotropia levator resection and lateral rectus resection were performed to repair what had been considered congenital ptosis. Additionally, median rectus resection was performed when the patient was 10 years old due to subsequent recurrent exotropia. There was no abnormality of the ECG at that time (Fig. 1B). He was diagnosed with type I diabetes and started insulin therapy at 15 years of age. At that time, the HbA1c level was 10.1%, and serum C-peptide levels were 0.221 ng/mg (fasting) and 1.19 ng/mg (postprandial).
At this time, CBC and ESR were within the normal ranges for the patient's age and sex. Serum lactate (25.2 mg/dl) and myoglobulin levels (150.6 ng/ml) were elevated. Liver function test showed mild elevation, with elevated AST (63 IU/L) and ALT (43 IU/L). Muscle enzymes lactate dehydrogenase (754 IU/L) and creatine phosphokinase (1139 IU/L) were also elevated. Other laboratory findings such as BUN (13.1 mg/dl), creatinine (0.93 mg/dl), Na (141 mEq/L), K (3.4 mEq/L), Ca (8.8 mg/dl), and Mg (2.0 mg/dl) were normal. Thyroid function test was also normal. Left ventricular ejection fraction was 61.1% without regional wall motion abnormality on echocardiography.
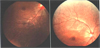
On ophthalmoscopy to discriminate KSS, the fundus showed atypical retinal pigmentary degeneration (Fig. 2). We impressed mitochondrial myopathy, and biopsy of the right thigh muscle (medial side) was performed. Unexpectedly, on light microscopic examination, modified trichrome stain revealed no diagnostic abnormalities such as ragged red fibers. Other enzyme histochemical and immunohistochemical stains were unremarkable (Fig. 3A), but electron microscopic examination, which is generally considered a more precise examination for mitochondria, revealed muscle cells with subsarcolemmal collections of mitochondria, many of them containing crystalline inclusions, electron microscopically consistent with mitochondrial myopathy (Fig. 3B).
After 3 days, ECG showed no recovery of normal sinus rhythm and persistent complete AV block, so we inserted a permanent pacemaker (DDDR type) (Fig. 1C). Cardiac MRI performed to exclude structural abnormalities before permanent pacemaker insertion showed normal cardiac chamber size and myocardial wall thickness and no myocardial delayed hyperenhancement, structural abnormality, or myocardial infiltrative disease.
We finally diagnosed Kearns-Sayre syndrome based on clinical manifestation, laboratory findings, and pathological confirmation.
DISCUSSION
Kearns-Sayre syndrome (KSS) has become recognized as a disease entity comprised of a constellation of features including progressive external ophthalmoplegia, pigmentary retinopathy, cardiac conduction system disorder, and multiple endocrine disorders.3
Muscle biopsy can confirm the diagnosis based on cytochrome C oxidase-negative ragged red fibers on modified Gomori stain. Electron microscopy reveals subsarcolemmal accumulation of abnormal mitochondria with paracrystalline inclusion bodies, and a more definite diagnosis based on mitochondrial DNA deletions determined by Southern blot or polymerase chain reaction should be sought.6
Our patient had several characteristic features of KSS, including progressive external ophthalmoplegia, retinal pigmentation, so-called “salt and pepper retinitis”, ptosis, muscle weakness, and mild cognitive disorders as well as type I diabetes (reported to affect 10% of KSS patients.3), metabolic acidosis, and high levels of lactate and creatine kinase. Of these, the cardiac manifestations, which create various degrees of AV block through the His-Purkinje system, are the most important factor for determining the prognosis of the disease.
In some rare cases, KSS has been diagnosed after death due to cardiac arrest. Most KSS patients have been diagnosed in Europe, the Middle East, and North America.4567 In Korea, one case involving the cardiac conduction system has been reported. That patient had a permanent pacemaker implanted, but the diagnosis was not confirmed by muscle biopsy.8
Our patient's diagnosis of KSS involving the cardiac conduction system was confirmed by muscle biopsy, which is rarely performed in Korea. In this case, we aimed to prevent sudden cardiac arrest by implanting a pacemaker at an early stage in patients who had specific ECG findings and it is meaningful to make follow up the KSS.
When we saw the KSS patients, we could detect right or left bundle branch block or complete AV block on ECG, Prophylactic pacemaker therapy is advisable in these patients because of the potential for progression of conduction abnormalities and risk of sudden death.


 XML Download
XML Download