

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Sjögren's syndrome is an inflammatory autoimmune disease characterized by lymphocytic infiltration of the lacrimal and salivary glands. The most common symptoms are keratoconjunctivitis sicca, xerostomia, and secretory gland-related symptoms such as parotid edema.
Among the symptoms found in regions other than the glands, the reported prevalence of cytopenia is as high as 30%. In a study conducted on 380 patients with Sjögren's syndrome, thrombocytopenia was found in 13% of individuals (62 patients), 0.4% of who showed a platelet count of 50,000/㎕ or less.1
Thrombocytopenia accompanied by an autoimmune disease is caused by autoantibody against the cell membranes, and it responds well to steroids in many cases. However, for those patients whose thrombocytopenia does not respond well to steroids, or in cases where thrombocytopenia recurs after reducing the dose of the administered steroid, combined administration with a high-dose steroid, immunosuppressant, and immunoglobulin is attempted.2
Here, the authors report a case of a patient who was diagnosed with Sjögren's syndrome during a differential diagnosis of thrombocytopenia and whose thrombocytopenia was addressed using a combined treatment of steroids and hydroxychloroquine, an antimalarial drug.
CASE
Patient: Ms. Park, a 52-year old.
History: The patient did not have a history of diabetes or high blood pressure, but she had a stroke which had occurred 7 year prior. She had been a smoker for 10 years. She has no family history of cardiovascular diseases, cerebrovascular diseases, or autoimmune diseases. She did not have a history of spontaneous abortion, but she had begun menopause 1 year earlier. She had not received estrogen replacement therapy or anticholinergic administration. The electrocardiography and angiocardiography performed at our institution 4.5 years earlier due to chest pain showed a non-ST-elevation myocardial infarction with proximal occlusion of the left circumflex artery. Following nonsegmented heparin medication and percutaneous coronary intervention, the patient was discharged and followed up with the administration of aspirin 100 ㎎, clopidogrel 75 ㎎, and rosuvastatin 10 ㎎ once a day. The blood test performed 2 years before the visit showed a decrease in platelet count from 70,000/㎕ to 90,000/µL (normal range from 140,000/㎕–440,000/㎕), and thus catamnesis was observed since then. Intermittent bruises and ecchymosis were observed a few months before the visit. The peripheral blood test performed 2 months prior to the visit showed a platelet level of 56,000/㎕ (normal range from 140,000–440,000/㎕) with normal prothrombin and activated partial thromboplastin times. Aspirin and clopidogrel were stopped and the patient was switched to sarpogrelate 100 ㎎ twice per day by substituting the antiplatelet drug administration. The follow-up test 1 month later showed a platelet level of 33,000/㎕, and the patient tested positive for antinuclear antibodies, and negative for antiplatelet antibodies. Hence, platelets were transfused and the sarpogrelate administration was discontinued.
Current case history: Due to the continued thrombocytopenia and positive antinuclear antibody findings, the patient was transferred to the Division of Rheumatology. Upon taking the patient's history, it was found that the patient had xeroma for more than 3 months and xerostomia with accompanying dental caries. In addition, the patient had Raynaud's phenomenon, alopecia, and photosensitivity, but did not show parotid edema, arthralgia, malar rash, discoid rash, canker sores, abdominal pain, abdominal discomfort, headaches, or lower limb edema or pain.
Physical findings: The physical examination showed a blood pressure of 110/70 mmHg, a heart rate of 66 beats/minute, a respiratory rate of 20 breaths/minute, a body temperature of 36.5℃, and a body mass index of 26.7 ㎏/㎡. Physical examination of the head showed neither parotid or major salivary gland enlargement, nor lymph node enlargement. Chest auscultation showed normal breath sounds in both lungs, and the heart sound was regular without noise. Abdominal palpation did not show liver or spleen enlargement, edema or tenderness in the lower limbs, or finger joint deformation or swelling. The skin exhibited livedo reticularis with no purpura, as well as mucosal hemorrhage.
Test findings: The peripheral blood test showed a hemoglobin level of 12.7 g/dL (normal range: 12–16 g/dL), a white blood cell count of 4,000/㎕ (normal range: 3,500–9,000/㎕), and a platelet count of 37,000/㎕ (normal range: 140,000–440,000/µL). The peripheral blood smear examination showed a normal white blood cell fraction without schistocytes and toxic granulation, and no giant thrombocytes were found. The test results also showed aspartate aminotransferase/alanine aminotransferase of 38/37 U/L (normal range: 10 to 45/10 to 45 U/L), a blood urea nitrogen of 10 ㎎/dL (normal range: 7–18 ㎎/dL), creatinine of 0.7 ㎎/dL (normal range: 0.6–1.2 ㎎/dL), and lactate dehydrogenase of 214 U/L (normal range: 140–271 U/L). The prothrombin and activated partial thromboplastin times were normal, and the direct Coombs test and the indirect Coombs test results were negative. The urine test showed no proteinuria, hematuria, pyuria, or bacteriuria.
The antibody tests were positive for antinuclear antibodies (1:80, homogeneous), negative for anti-double-stranded deoxyribonucleic acid immunoglobulin (Ig)G antibody, negative for anti-Sm antibody, negative for ribonucleoprotein (RNP) antibody, negative for anti-La antibody, and positive for anti-Ro antibody. The tests also showed negative lupus anticoagulant factor antibody, negative anticardiolipin IgM and IgG antibody, C3 103 ㎎/dL (normal range: 90–180 ㎎/dL), C4 18 ㎎/dL (normal range: 10–40 ㎎/dL), negative anti-beta-2-glycoprotein I IgM and IgG antibody, and negative anti-Scl 70 antibody. The patient was negative for anti-HCV and human immunodeficiency virus. The cancer marker test results were as follows: carcinoembryonal antigen 2.2 ng/mL (normal range: 0–2.5 ng/mL), CA19-9 25.9 U/mL (normal range: 0–37 U/mL), and CA125 81.0 U/mL (normal range: 0–35 U/mL), which was high, but it was later found to be 13.5 U/mL (in the normal range) in the follow-up test performed 2 months later. Abdominal ultrasonography did not show spleen enlargement. No abnormalities were found in either pulmonary parenchyma by plain chest radiography.
The result of Schirmer's test performed for the differentiation of Sjögren's syndrome was negative. However, salivary gland scanning showed decreased absorption of the right parotid gland and the salivary gland of the right jaw. Mucosal biopsy of lower lip was not performed since the patient did not consent to this.
Treatment and catamnesis: According to the international classification criteria for Sjögren's syndrome, which were revised in 2002, the patient was diagnosed with primary Sjögren's syndrome on the basis of the presence of xeroma with discomfort for 3 months or longer, xerostomia accompanying dental caries, the positive scan findings of the salivary gland, and the positive anti-Ro antibody finding without identification of another autoimmune disease. Pilocarpine hydrochloride was administered for xerostomia, while prednisolone 10 ㎎/day was orally administered for thrombocytopenia. Then, intermittent bruises were continuously found around the body. Since the regular peripheral blood test showed a decrease in the platelet count down to 35,000/㎕, the dosage of prednisolone was increased to 20 ㎎/day from the 9th month of steroid treatment.
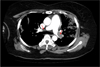
One year and five months after the start of the steroid treatment, while the patient was being administered 7.5 ㎎/day of prednisolone, and the platelet level was maintained at a range from 30,000–69,000/㎕, the patient visited our institution with a chief complaint of respiratory distress. Chest computed tomography (CT) showed pulmonary thromboembolism (Fig. 1), and the follow-up test performed after hospitalization in division of pulmonology showed negative antiplatelet antibody, negative lupus anticoagulant factor antibody, negative anticardiolipin IgM and IgG, and negative anti-beta-2-glycoprotein I IgM and IgG antibodies. For the pulmonary thromboembolism, enoxaparin 1 ㎎/㎏ was hypodermically injected twice per day and warfarin 5 ㎎ was orally administered. The outpatient chest CT performed 6 months after the start of the anticoagulant treatment showed that the pulmonary thromboembolism was completely recovered, and thus anticoagulant treatment was discontinued.
Two years and five months after starting the steroid treatment, the administration of prednisolone 12.5 ㎎/day was maintained, but the platelet level was decreased down to 30,000/㎕. Since the steroid dosage could not be reduced, oral administration of hydroxychloroquine 300 ㎎/day was additionally performed. Four months after starting the combined administration of hydroxychloroquine, the patient's platelet level increased to 74,000/㎕. The prednisolone dosage was gradually decreased after maintaining the hydroxychloroquine therapy at 300 ㎎/day. Fifteen months after the start of the combined administration of hydroxychloroquine, the patient's platelet level was recovered to 106,000/µL without any adverse drug effects or exacerbation of the xerostomia or xeroma. The prednisolone dosage was reduced to 2.5 mg/day and a platelet level of 60,000–100,000/㎕ has been maintained (Fig. 2).
DISCUSSION
The patient in the present case report was treated with nonsegmented heparin and antiplatelet drug medication due to a myocardial infarction that was diagnosed 4.5 years before her visit to our institution. However, even after the medication was discontinued, the platelet level was not recovered. Since no drugs that cause thrombocytopenia were included in the patient's medication, drug-induced thrombocytopenia was empirically excluded. The patient's medical history showed myocardial infarction that was definitively diagnosed by angiocardiography, and the patient was diagnosed with pulmonary thromboembolism during the treatment of thrombocytopenia. Therefore, the clinical criteria for antiphospholipid antibody syndrome, firstly presented in Japan in 1998 and revised in 2006, were satisfied. However, the results from the lupus anticoagulant factor antibody tests, the anticardiolipin IgG/IgM isotype test, and the anti-beta-2-glycoprotein I IgM and IgG isotype test performed at an interval longer than 12 weeks were all negative, thus not satisfying the classification criteria for antiphospholipid antibody syndrome. The patient was diagnosed with primary Sjögren's syndrome due to the presence of eye symptoms, oral symptoms, salivary gland infiltration verified by the positive findings of the salivary gland scan, and the positive anti-Ro antibody finding, which satisfied the classification criteria for Sjögren's syndrome that was jointly proposed by the United States and Europe in 2002.
Autoimmune diseases such as systemic lupus erythematosus and rheumatoid arthritis are known to increase the risk of cardiovascular diseases, as they involve atherosclerosis with traditional risk factors. A retrospective cohort study conducted in 2015 with 1,343 primary Sjögren's syndrome patients showed that the primary Sjögren's syndrome increases the risk of cerebrovascular diseases and myocardial infarction.3
A recent retrospective cohort study conducted with 8,920 Sjögren's syndrome patients, using Taiwan's National Health Insurance Research Database (NHIRD), showed that the patients had a pulmonary thromboembolism incidence rate that was 3.29 higher than that of the control group, and it was reported that Sjögren's syndrome itself is an independent risk factor.4 The patient in the present case report had neither a family history of early coronary artery diseases, nor a medical history of diabetes, hyperlipidemia, or hypertension. However, the patient experienced cerebral infarction 7 years prior to the visit and myocardial infarction 4.5 years prior to the visit; then, pulmonary thromboembolism occurred 1.5 years after starting a steroid treatment for thrombocytopenia. Although the patient had traditional risk factors such as 10-year smoking history, the use of steroids, and the onset of menopause, thromboembolism was found three times in different organs, which is unusual given the patient's relatively young age, suggesting that the patient had Sjögren's syndrome, an autoimmune disease. To verify the involvement of Sjögren's syndrome, it is necessary to conduct further studies; domestic epidemiological surveys focused on the correlation between Sjögren's syndrome and thromboembolism with respect to the biological mechanisms, risk factors, predictive factors, and prevention.
Cytopenia, which is the most common symptom of primary Sjögren's syndrome, has a prevalence rate of 14%–42% as leukocytopenia, about 11% as anemia, and 5%–15% as thrombocytopenia.5
Thrombocytopenia is defined as a platelet level lower than the lowest 2.5th percentile in the normal platelet number distribution. The 3rd U.S. National Health and Nutrition Examination Survey (NHANES Ⅲ) stated that a platelet level of 150,000/㎕ is the appropriate normal lower limit.
Thrombocytopenia without an abnormality of the red blood cells and white blood cells, and without systemic symptoms, is defined as isolated thrombocytopenia, of which most common cause is idiopathic thrombocytopenic purpura and drug-induced thrombocytopenia. Additionally, isolated thrombocytopenia may be caused by infection with human immunodeficiency virus or hepatitis C virus, and by antiphospholipid antibody syndrome in the case of a connective tissue disease such as systemic lupus erythematosus and rheumatoid arthritis.6
Although severe thrombocytopenia requiring treatment in Sjögren's syndrome patients has been reported, thrombocytopenia is mostly mild and shows a symptomless catamnesis for a few years. Thrombocytopenia in Sjögren's syndrome may be isolated or accompanied by another type of cytopenia. Therefore, a diagnosis of Sjögren's syndrome with a chief complaint of thrombocytopenia may be late to detect.7
In the present case, the patient intermittently showed thrombocytopenia in the range of 70,000–90,000/µL in blood tests that were performed about 2 years prior to the patient's diagnosis of Sjögren's syndrome. This suggests that it is important to consider an autoimmune disease as a differential disease during the patient's initial history taking, physical examination, and blood tests when a patient has thrombocytopenia, and it is also essential to verify the symptoms and signs that imply an autoimmune disease in order to make a timely diagnosis.
Although the accurate mechanism has yet to be determined, it is known that immune thrombocytopenia destroys platelets through autoantibodies or immunocomplexes against the glucoproteins in cellular walls. A study published in 2007, where the medical records of 125 Sjögren's syndrome patients were retrospectively investigated, showed that decreased C4 and positive antinuclear antibodies are significantly correlated with thrombocytopenia accompanied by Sjögren's syndrome.8 Steroids are used as a primary treatment for immune thrombocytopenia, a treatment that is effective in 50%–80% of patients. The pharmacological mechanism responsible for the effect of steroids is known to be the acceleration of apoptosis, the inhibition of T-lymphocyte activation, and the suppression of B-lymphocyte differentiation. The long-term use of steroids may cause side effects such as hypertension, diabetes, osteoporosis, and adrenalism, whereas a decreased steroid dosage may exacerbate thrombocytopenia and an discontinued steroid dosage may cause a low remission rate (10%–30%). Therefore, efforts are made to find a drug that can be administered in combination with conventional steroids, or even that can replace steroids. Although immunosuppressors such as mycophenolate mofetil and azathioprine, immunoglobulins, and high-dose steroids showed a treatment effect in some cases, they were associated with drug side effects and recurrence.2
The patient in the present case did not have a severe hemorrhage, except for intermittent bruises and ecchymosis; her platelet level did not decrease below 30,000/㎕. However, a low-dose steroid treatment was performed given the presence of risk factors for thrombosis, including uncorrected obesity, smoking, menopause, cerebral infarction that occurred at a relatively young age, and the history of a myocardial infarction. The patient was treated with prednisolone, and her platelet levels were monitored for 1.5 years following the diagnosis; however, the dose could not be reduced be low 12.5 mg/day. Therefore, a combined or alternative treatment was taken into consideration. The literature review showed two case reports of overlap syndrome patients having systemic lupus erythematosus and Sjögren's syndrome, where thrombocytopenia continued and a low blood 25-hydroxy vitamin D concentration was found despite steroid treatment (prednisolone 40–60 ㎎/day); however, the patients improved following the use of combined hydroxychloroquine 400 ㎎/day and high-dose vitamin D with steroid.2 When applying the medication in these cases, hydroxychloroquine 300 ㎎/day was combined with prednisolone 12.5 ㎎/day. Fifteen months after the start of the combined medication, the patients' platelet levels improved to 106,000/㎕, and thus the dose of prednisolone could be gradually reduced to 2.5 ㎎/day.
The accurate mechanism of hydroxychloroquine has not been clearly identified, but hydroxychloroquine is known to have various immunomodulatory effects such as lysosomal acidification, phagocytosis, antigen presentation, cytokine secretion mediated by macrophages, and suppression of calcium signal transduction through B-cell and T-cell receptors. Given that it was recently found that hydroxychloroquine has anticancer, antithrombotic, and antibiotic effects, the scope of hydroxychloroquine application is thus being extended.9 In addition, since hydroxychloroquine inhibits platelet agglutination and arachidonic acid secretion, and it also suppresses the action of antiphospholipid antibodies, it is used to prevent thromboembolism in patients who are hard to move and antiphospholipid syndrome patients.9 By administering hydroxychloroquine to the patient in the present case (who has a history of thromboembolism), it may have a dual effect insofar as it not only treats thrombocytopenia, but it also prevents thromboembolism.
In one study, the use of hydroxychloroquine (200–600 ㎎/day) as a secondary drug was administered in 28 immune thrombocytopenia patients with positive antinuclear antibodies whose conditions did not improve despite the long-term administration of steroids, immunoglobulins, and immunosuppressors as primary drugs. In that study, most of the patients showed an improved platelet level as early as 3 months after the start of the combined medication without any side effects associated to the drugs, which was the result of the delayed onset pharmacological action of hydroxychloroquine. Therefore, the authors stated that a drug combination involving hydroxychloroquine is needed from the early stages of treatment.10 In the present case report, the patient's platelet level was improved as soon as 4 months after the start of the combined administration of hydroxychloroquine and prednisolone, without any side effects such as visual field defects, nausea, vomiting, abdominal pain, headache, fatigue, and nervousness, and thus the prednisolone dosage could be effectively reduced. Although further studies are needed to investigate the mechanism of action, as well as the efficacy, stability, proper dose, and administration period, the possibility of using hydroxychloroquine as a secondary drug for the treatment of thrombocytopenia accompanied by Sjögren's syndrome was verified in the present case study, as well as in previous studies. Sufficient combined treatment for 3 months or longer, as well as treatment response assessment, may be necessary.

 XML Download
XML Download