

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Cerebral ischemia causes severe neurologic deficits and impairments, consequently leading to movement disorders and cognitive impairment [12]. Cerebral ischemia is involved with various biochemical processes such as the generation of reactive oxygen species and excessive nitric acid, along with the activation of the inflammatory response and excitatory neurotransmitters [345]. These changes trigger neuronal degeneration and neuronal cell death, consequently leading to severe neuronal damage and neuronal dysfunction [6].
The synuclein protein family has three members: α-, β-, and γ-synuclein, which are primarily found in neuronal tissue. Among these members, α-synuclein is a ubiquitous protein that is expressed abundantly in the brain. It is especially localized in the presynaptic terminals of neurons and involved in synaptic vesicle regulation [7]. α-Synuclein directly inhibits lipid oxidation by acting as an antioxidant through binding to a lipid membrane, which allows the four methionine residues of α-synuclein to become oxidized to sulfoxide by hydrogen peroxide [89]. Lipid peroxidation is a pathogenic process in focal cerebral ischemic injury [10]. Reactive oxygen species lead to lipid peroxidation that consequentially triggers damage in the plasma and intracellular membrane [1011]. The mediation of α-synuclein is closely related with lipid peroxidation in neuronal cells [12]. Thus, we propose that there are changes in α-synuclein expression in ischemic brain injury. In this study, we investigated α-synuclein expression in both a cerebral ischemic animal model and neuronal cells damaged by glutamate treatment.
Materials and Methods
Experimental animals
Male Sprague-Dawley rats weighing 200-220 g (n=30) were obtained from Samtako Co. (Animal Breeding Center, Osan, Korea). Rats were divided into two groups, sham-operated and middle cerebral artery occlusion (MCAO) animals (n=15 per group). Animals were maintained under controlled light (12 hours light/12 hours dark cycle) and temperature (25℃) conditions. All experimental procedures on animals were conducted according to the approved guidelines of the Institutional Animal Care and Use Committee of Gyeongsang National University (GNU-LA-015).
Middle cerebral artery occlusion
MCAO surgical operation was performed to induce focal cerebral ischemia as previously described [13]. For anesthetization, animals were administrated Zoletil (50 mg/kg; Virbac, Carros, France) via intramuscular injection. After a midline incision was given in the neck, right common carotid artery, external carotid artery, and internal carotid artery were separated from adjacent nerves and muscles. Suprathyroid and occipital arteries were ligated, and then followed ligation of the external carotid and pterygoid arteries. The right common carotid artery was then clamped using microvascular clips. A small incision was made in the external carotid arteries, and a 4/0 monofilament nylon with a blunted tip was inserted intraluminally and advanced into the internal carotid artery to occlude the middle cerebral artery. The external carotid artery containing the monofilament nylon was ligated using black silk, the microvascular clips on the right common carotid artery were removed, and the neck incision was competently sutured. Animals were sacrificed and brain tissues were collected 24 hours after the onset of occlusion. Animals that underwent the same surgical process except insertion of the nylon were used as sham-operated animals.
Neurological behavioral test
Neurological function tests were performed according to a previously described manual [13]. Neurological function was evaluated using a five-point scoring system: no neurologic deficit (0), mild focal neurologic deficit with failure to fully extend the left forepaw (1), moderate focal neurologic deficit with occasional rotating movement (2), severe focal deficit with spontaneous circling (3), severe deficit with no spontaneous movement and depressed consciousness (4).
TTC staining
Brain tissues were coronally sectioned at a thickness of 2 mm using a Brain Matrix (Ted Pella, Redding, CA, USA). For histological staining, brain slices were incubated in 2% triphenyltetrazolium chloride (TTC; Sigma, St. Louis, MO, USA) solution at 37℃ for 20 min. The stained tissues were then fixed in 10% formalin and scanned by an Agfa ARCUS 1200™ (AgfarGevaert, Mortsel, Belgium). Infarct volumes were evaluated using Image-ProPlus 4.0 software (Media Cybernetics, Silver Spring, MD, USA). The percentage of the ischemic area was calculated by the following formula: infarction area/whole slice area ×100.
Histopathological study
Brain slices fixed in formalin were washed with tap water, dehydrated in a gradient of ethanol from 70% to 100%, and cleared in xylene. Brain slices were embedded with paraffin in an embedding center with a vacuum pump, and the paraffin blocks were sectioned into 4 µm coronal sections using a microtome (Leica, Wetzlar, Germany). Paraffin ribbons were put on glass slides, dried on a slide warmer (Fisher Scientific, Hampton, NH, USA), deparaffined in xylene, rehydrated in a gradient of ethanol from 100% to 70%, and stained with hematoxylin and eosin solution (Sigma) according to the general staining method. After staining, dehydrated tissue sections were mounted in permount medium (Sigma) and observed using a light microscope.
Two-dimensional gel electrophoresis
The right cerebral cortexes of the rats were homogenized in lysis buffer solution (8 M urea, 4% CHAPS, ampholytes, and 40 mM Tris-HCl), and the homogenates were centrifuged at 16,000×g for 20 min at 4℃. After centrifugation, the supernatant was discarded, and the pellet was resuspended in lysis buffer for protein extraction. The total protein concentration was determined by the Bradford protein assay kit (Bio-Rad, Hercules, CA, USA) according to the manufacturer's instructions. Total protein samples were separated using two-dimensional gel electrophoresis. First-dimensional isoelectric focusing (IEF) was performed on an Ettan IPGphor 3 System (GE Healthcare, Uppsala, Sweden) using immobilized pH gradient (IPG) gel strips (17 cm, pH 4-7 and pH 6-9; Bio-Rad). IPG strips were rehydrated in sample buffer (8 M urea, 2% CHAPS, 20 mM DTT, 0.5% IPG buffer, and bromophenol blue) that contained protein samples (100 µg) for 13 hours before IEF. After rehydration, IEF was carried out as follows: 250 V for 15 min, 10,000 V for 3 hours, followed by an increase from 10,000 V to 50,000 V. Subsequently, IPG strips were incubated with equilibration buffer [6 M urea, 30% glycerol, 2% SDS, 50 mM Tris-HCl (pH 8.8), 1% DTT] for 15 min. Second dimension electrophoresis was performed to separate protein samples based on molecular weight. Strips were positioned on gradient gels (7.5-17.5%) for loading. The gels were installed in Protein-II XI electrophoresis equipment (Bio-Rad) and loaded until bromophenol blue reached the bottom side of the gel using the following conditions: 5 mA for 2 hours and 10 mA for 10 hours at 10℃.
Silver staining, image analysis, and protein identification
Gels were incubated with fixation solution (12% acetic acid, 50% methanol) for 2 hours and treated with 50% ethanol for 20 min. After fixation, the gels were incubated with silver staining solution (0.2% silver nitrate) for 20 min to stain the gels and then developed in a 0.2% sodium carbonate solution. Stained gels were scanned with an Agfa ARCUS 1200 in an image format (Agfa-Gevaert, Mortsel, Belgium). PDQuest 2-D analysis software (Bio-Rad) was employed to analyze protein spots. Specific protein spots were cut from the gels, gel particles were digested with trypsin-containing buffer, and proteins were extracted for MALDI-TOF. Peptides of significance were investigated using the Voyager System DE-STR MALDI-TOF mass spectrometer (Applied Biosystems, Foster City, CA, USA). MS-Fit and ProFound programs were applied to detect proteins, and protein sequences were identified from the SWISS-Prot and NCBI databases.
Cell culture and glutamate treatment
HT22 cells derived from a mouse hippocampal cell line were used for the in vitro experiment. HT22 cells were maintained in Dulbecco's modified Eagle's medium (DMEM, without L-glutamate, Gibco BRL, Gaithersburg, MD, USA) containing 10% fetal bovine serum, penicillin (100 units/mL), and streptomycin (100 µg/mL). Cells were incubated under proper conditions (37℃, 5% CO2 atmosphere). For the cell viability assay, HT22 cells were seeded at 5,000 cells per 100 µL DMEM media in each well of a 96-well plate and cultured overnight. Cells were also seeded on 60-mm culture dishes at 100,000 cells per dish for further studies. Cell density was maintained at 70% confluency or less and was monitored to prevent excessive growth as described previously [14]. The medium was replaced with different concentrations of glutamate (Sigma, 1, 3, or 5 mM)-treated medium 24 hours after cell seeding. Cells were collected after 24 hours of glutamate treatment.
MTT assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed to measure cell viability. The MTT solution (5 mg/mL) was added to each well, and cells were incubated at 37℃ for 4 hours. After discarding the MTT solution, a solubilization solution (1 mL) containing 20% sodium dodecyl sulfate (pH 4.8) and 50% dimethylformamide was added to the medium, and the cells were incubated overnight. The absorbance of the solubilization solution was measured at a wavelength of 570 nm. Cell viability was represented as a ratio vs. vehicle set at 1.
Western blots
Brain tissue was homogenized with lysis buffer (1% Triton X-100, 1mM EDTA in 1× PBS, pH 7.4) containing 200 µM phenylmethylsulfonyl fluoride. Homogenates were centrifuged at 15,000×g for 20 min at 4℃, and the supernatants were isolated. The bicinchoninic acid protein assay kit (Pierce, Rockford, IL, USA) was used to measure the protein concentration of the supernatant. Protein samples (30 µg) were mixed with gel-loading buffer, denatured at 100℃, and cooled on ice. Protein was loaded into each well for 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). For minimization of nonspecific antibody binding, membranes were blocked in 5% skim milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 hour and washed three times with TBST for 10 min each. The membranes were incubated with the primary antibodies anti-α-synuclein (1:1000, Cell Signaling Technology, Beverly, MA, USA) and anti-actin (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4℃. After washing with TBST, the membranes were incubated with the following secondary antibodies: horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG (1:5000, Cell Signaling Technology) for 2 hours. Subsequently, the membranes were washed with TBST, reacted with enhanced chemiluminescence detection reagents (GE Healthcare, Little Chalfont, Buckinghamshire, UK) for detection of immuno-reactive protein signals according to the manufacturer's protocol, and exposed to X-ray film (Fujimedical X-ray film, FujiFilm) and developed.
Data analysis
All data are expressed as mean±SEM. Intensity analysis was carried out using SigmaGel 1.0 (Jandel Scientific, San Rafael, CA, USA) and SigmaPlot 4.0 (SPSS Inc., Point Richmond, CA, USA). The results for each group were compared by one-way analysis of variance (ANOVA) followed by Student's t-test. Differences in comparisons were considered significant at P<0.05.
Results
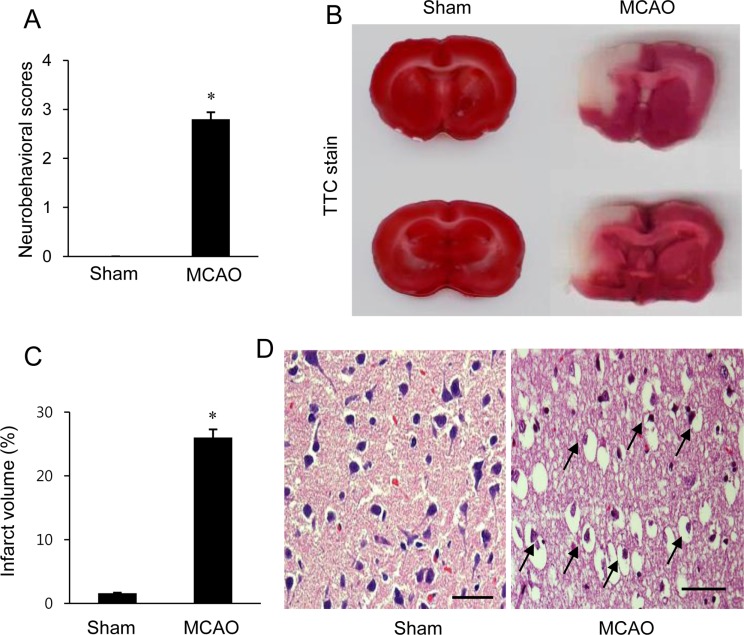
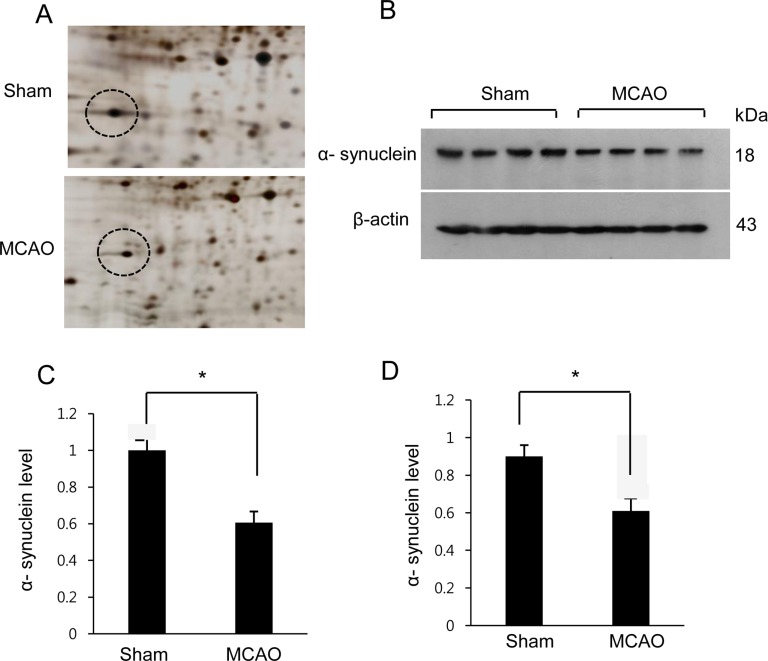
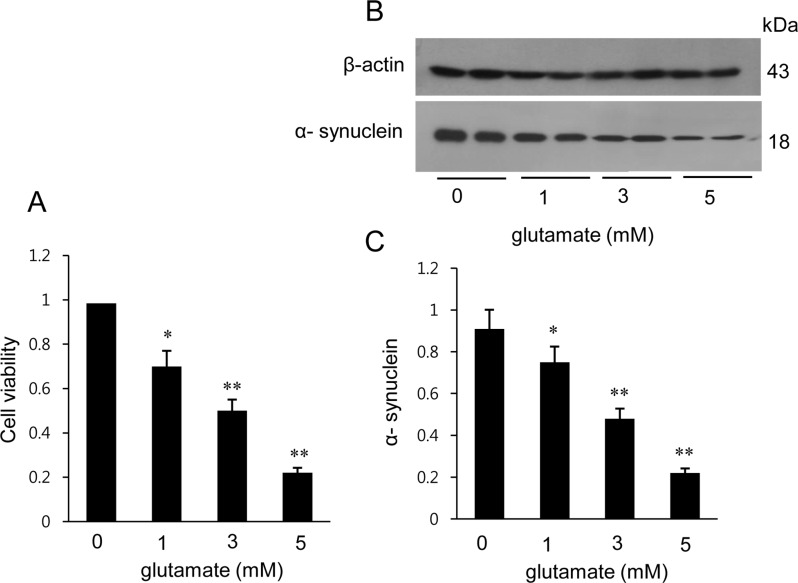
We confirmed that MCAO-induced ischemic injury leads to neurological behavioral deficit and cerebral cortex damage. MCAO-operated animals showed neurological deficit ranging from occasional rotating movements to spontaneous circling (neurological function scores of 2 to 3), while obvious signs of neurological deficits were not observed in sham-operated animals (Figure 1A). Moreover, TTC staining clearly showed an increase in infarct volume in MCAO-operated animals (Figure 1B). Infarct volumes were 1.9±0.3 and 24.8±2.7% in sham-operated and MCAO-induced animals, respectively (Figure 1C). We also observed histopathological changes in the cerebral cortex of MCAO-operated animals. Most neuronal cells in the brain tissue from MCAO animals appeared shrunken and swollen in shape and had nuclei with apoptotic bodies containing intensive dark masses, while intact neurons with a pyramidal shape were observed in sham-operated animals (Figure 1D). We observed a decrease in α-synuclein expression in MCAO-induced animals using a proteomic technique (Figure 2A). The peptide mass of α-synuclein is 6/134 and the sequence of this protein is 43%. α-Synuclein levels were 1.02±0.03 and 0.58±0.02 in the cerebral cortices of sham-operated and MCAO-induced animals, respectively (Figure 2C). Western blot analysis also clearly demonstrated a decrease in α-synuclein expression in MCAO-induced animals compared to sham-operated animals (Figure 2B). α-Synuclein protein levels were 0.91±0.03 and 0.63±0.05 in the cerebral cortices of sham-operated and MCAO-induced animals, respectively (Figure 2D). In the in vitro experiment, glutamate treatment in cultured HT22 cells decreased cell viability in a dose-dependent manner and induced neuronal cell death (Figure 3A). Moreover, Western blot analysis showed that glutamate toxicity induced a decrease in α-synuclein level in a dose-dependent manner (Figure 3B). α-Synuclein level was 0.92±0.05 in the vehicle-treated group and decreased to 0.75±0.03, 0.47±0.04, and 0.28±0.03 in the 1, 3, and 5 mM glutamate-treated groups, respectively (Figure 3C).
Discussion
The MCAO surgical operation is used to induce cerebral ischemia that leads to infarct lesions and neuronal cell damage in a representative animal model [1315]. Cerebral ischemia leads to neuronal cell death through the up- and down-regulation of various proteins [1516]. This study demonstrated that there is a decrease in the expression of α-synuclein protein in MCAO-induced brain injury. We confirmed that the MCAO operation increases infarct volume and apoptotic cell death, consequently causing histopathological changes in the cerebral cortex and neurological behavior deficits. This study focused on α-synuclein expression in the cerebral cortex following MCAO and glutamate-exposed hippocampal neurons.
Many studies have shown that mutations in the α-synuclein gene are related to neurodegenerative disorders such as Parkinson's disease and Alzheimer's disease [171819]. α-Synuclein also plays a critical role in the pathogenesis of neurodegenerative diseases. This study showed a decrease in α-synuclein after MCAO injury using a proteomic approach and Western blot analysis. A previous study demonstrated that α-synuclein expression prevented neuronal cell death induced by hydrogen peroxide or serum withdrawal [20]. Moreover, α-synuclein attenuates the reduction in glutathione after serum deprivation and hydrogen peroxide treatment in neuronal cells [20]. Glutathione plays an important role as an antioxidant, anti-aging agent, and lipid oxidative agent. It is well known that glutathione can attenuate the cellular damage caused by reactive oxygen species such as free radicals, lipid peroxides, and peroxide [21]. Mutant α-synuclein increases lipid peroxidation and protein carbonyls, consequently accelerating cell death in response to toxic insults [20]. Our results reported a decrease in α-synuclein after cerebral ischemia, which has been shown to be related with lipid peroxidation and neuronal cell death. Moreover, α-synuclein contributes to neuronal survival through the PI3/Akt kinase pathway and exerts neuroprotective effects through regulation of expression of the Bcl-2 protein family [22]. In addition to observing a decrease in α-synuclein following brain ischemic injury in an animal model, we also examined changes in α-synuclein in neurons exposed to glutamate through an in vitro study. Glutamate toxicity induces the generation of excessive reactive oxygen species and mitochondrial dysfunction and consequently leads to neuronal cell death [14]. We confirmed a dose-dependent decrease in both cell viability and α-synuclein level in glutamate-exposed hippocampal cells. Decreases in α-synuclein following glutamate exposure indicate neuronal damage. We observed a decrease in α-synuclein in both an MCAO-induced injury animal model and in glutamate-exposed cultured neurons, which suggests that the reduction in α-synuclein is mediated by neuronal cell death following brain ischemic injury.
 XML Download
XML Download