

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Stroke is a major cause of death and 87% of all stroke cases are cerebral ischemic strokes. Cerebral ischemic injury induces intracellular calcium overload, mitochondrial dysfunction, reactive oxygen species (ROS) generation, inflammation, and excitotoxicity [12]. This consequently leads to serious neuronal cell damage, disruption of neuronal function, and adult disability. Mitochondria are involved in several functions including cellular energy production, signaling, differentiation, growth, and death [3]. The conditions resulting from a stroke lead to mitochondria dysfunction [4].
Dynamin-like protein 1 (DLP-1) is an essential mitochondrial fission and fusion protein that is mainly localized in synapses [5]. The DLP-1 protein, a GTPase protein, is closely implicated in mitochondrial division, distribution, and other dynamic actions [67]. The imbalance between mitochondrial fission and fusion leads to structural impairment and dysfunction in mitochondria. Consequent abnormalities in mitochondria function cause the disruption of ATP production and neuronal damage [8910]. DLP-1, which is activated by Bcl-xL, induces synaptic formation in hippocampal neurons [11]. In addition, DLP-1 knockout mice exhibit developmental abnormalities in the forebrain due to deficient synaptic formation and mitochondria aggregation [1213]. Therefore, DLP-1 is a crucial protein for the regulation of mitochondrial and synaptic functions underlying neuronal damage. However, little information is available regarding change in DLP-1 expression after ischemic brain injury. In this study, we examined DLP-1 expression in ischemic injury and investigated DLP-1 expression in a focal cerebral ischemic animal model as well as in neuronal cells with glutamate-induced damage.
Materials and Methods
Experimental animals and middle cerebral artery occlusion
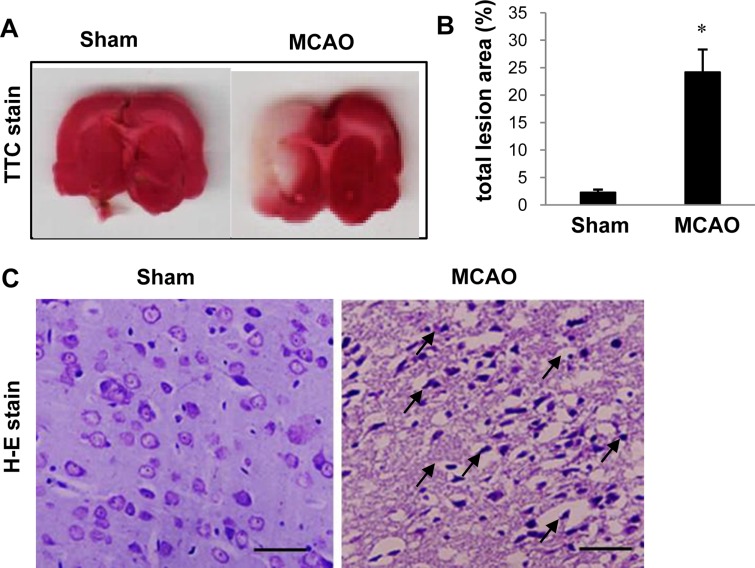
Male Sprague-Dawley rats (210-220 g, n=30) were obtained from Samtako Co. (Animal Breeding Center, Osan, Korea) and were divided into two groups, sham-operated and middle cerebral artery occlusion (MCAO) animals (n=15 per group). Rats were allowed free access to food and water, and were kept in a temperature and light-controlled environment. All experimental procedures for animal use were approved by the Institutional Animal Care and Use Committee of Gyeongsang National University (GNU-LA-015). To induce focal cerebral ischemia, MCAO was surgically performed using a previously described method [14]. Rats were anesthetized with Zolretil (50 mg/kg, Virbac, Carros, France) before the MCAO operation. The right common carotid artery, external carotid artery, and internal carotid artery were exposed through a neck midline incision. A 4/0 nylon monofilament with a heated round tip was introduced into the right external carotid artery and advanced into the internal carotid artery until the tip blocked the origin of the middle cerebral artery. Sham-operated rats underwent the same surgical procedure without the insertion of the nylon monofilament. Animals were kept on a heating pad to maintain body temperature. Twenty-four hours after blocking the middle cerebral artery, the brains were removed and cut into 2 mm thick coronal slices. The brain slices were stained in 2% triphenyltetrazolium chloride (TTC; Sigma, St. Louis, MO, USA) at 37℃ for 20 min and fixed in 10% formalin. For the histopathological study, the brain slices were embedded with paraffin and the paraffin block were cut into 4 µm coronal section. The paraffin sections were deparaffined in xylene, rehydrated in gradient ethanol from 100% to 70%, and stained with hematoxylin and eosin solution (Sigma). The dehydrated tissue sections were mounted with permount (Sigma) and observed under a light microscope.
Two-dimensional gel electrophoresis, image analysis, and protein identification
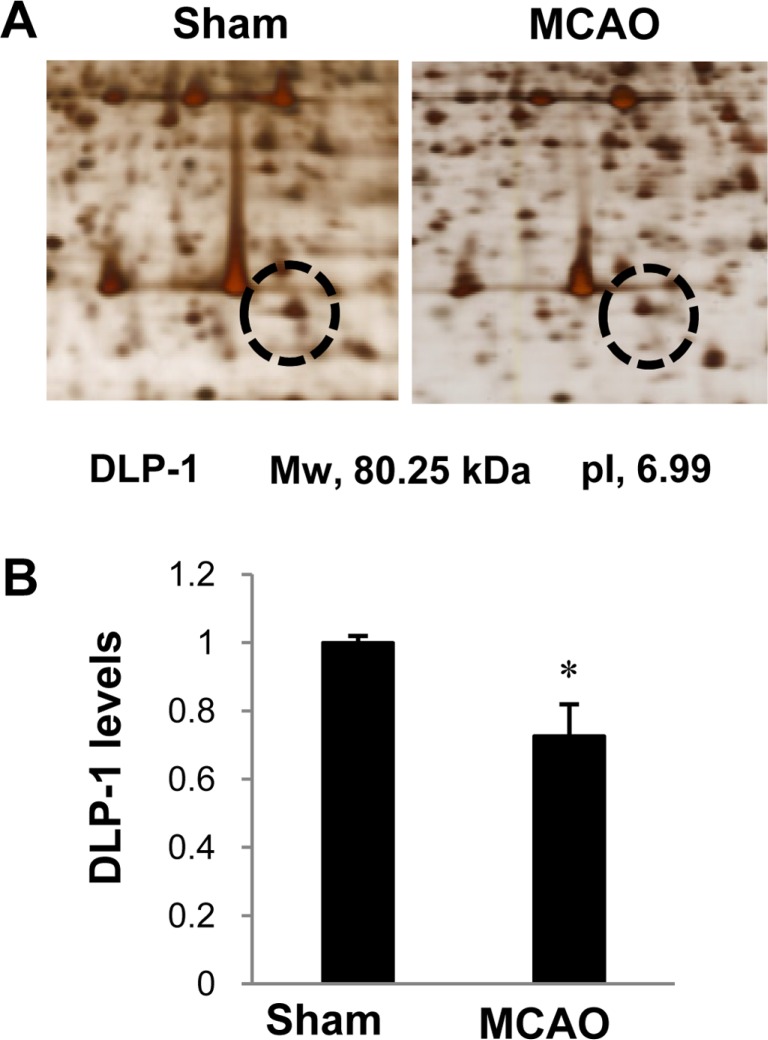
The proteomics study was performed using a previously described method [15]. Twenty-four hours after MCAO, the right cerebral cortices were isolated, homogenized in lysis buffer (8 M urea, 4% CHAPS, ampholytes, and 40 mM Tris-HCl), and centrifuged at 16,000 g for 20 min at 4℃. After centrifugation, the pellets were dissolved in lysis buffer and the protein concentration was measured using the Bradford method (Bio-Rad, Hercules, CA, USA) according to the manufacturer's protocol. The Ettan IPGphor 3 System (GE Healthcare, Uppsala, Sweden) was used for first-dimensional isoelectric focusing (IEF). Immobilized pH gradient (IPG) gel strips (17 cm, pH 4-7 and pH 6-9; Bio-Rad) were rehydrated in buffer (8 M urea, 2% CHAPS, 20 mM DTT, 0.5% IPG buffer, and bromophenol blue) for 13 hours. The protein samples (100 µg) were loaded onto the IPG strips using a sample cup and IEF was performed as previously described [15]. The strips were subjected to gradient gels (7.5-17.5%) for sodium dodecyl sulfate (SDS) gel electrophoresis. The gels were loaded on Protein-II XI electrophoresis equipment (Bio-Rad) and subjected to a 5 mA for 2 hours, followed by 10 mA for 10 hours at 10℃. The gels were fixed in 12% acetic acid and 50% methanol for 2 hours and washed with 50% ethanol for 20 min. The gels were stained with silver solution (0.2% silver nitrate) for 20 min and developed in a solution of 0.2% sodium carbonate. The gel images were scanned with an Agfa ARCUS 1200™ (Agfa-Gevaert, Mortsel, Belgium). PDQuest 2-D analysis software (Bio-Rad) was used for analysis of the protein spots. Protein spots were excised from the gels and processed for matrix assisted laser desorption ionizationtime of flight (MALDI-TOF). The gel particles were digested in trypsin-containing buffer and extracted. Peptides were analyzed with a Voyager System DE-STR biospectrometry workstation (Applied Biosystem, Foster City, CA, USA). MS-Fit and ProFound programs were used to detect proteins and the databases SWISS-Prot and NCBI were used to identify protein sequences.
Western blot analysis
The right cerebral cortices were homogenized in lysis buffer [1% Triton X-100, 1 mM EDTA in PBS (pH 7.4)] and centrifuged at 15,000 g for 20 min at 4℃. After centrifugation, the supernatants were harvested for further processing and the protein concentration was measured according to the manufacturer's protocol. From each sample, 30 µg of total protein was loaded on 10% SDS polyacrylamide gel and electrophoresis was performed. The electrophoresed proteins were transferred onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA) and the membranes were washed in Tris-buffered saline containing 0.1% Tween-20 (TBST). The membranes were blocked with 5% milk solution for 1 hour to minimize non-specific antibody binding and incubated with antibodies against anti-DLP-1 (diluted 1:1,000, BD Biosciences, San Jose, CA, USA) and antiactin (diluted 1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) as the primary antibodies. The membranes were washed with TBST and reacted with horseradish perxoxidase-conjugated goat anti-rabbit IgG secondary antibody (1:5,000; Santa Cruz Biotechnology). The enhanced chemiluminescence Western blot analysis system (Amersham Pharmacia Biotech, Piscataway, NJ, USA) was used for the detection of immunoreactive bands.
Cell culture and glutamate treatment
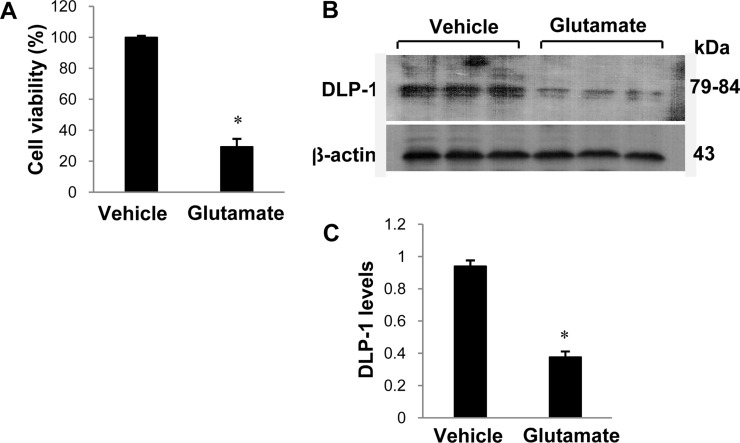
The cell culture system was performed used as previously described [16]. A mouse hippocampal cell line (HT22) was grown in Dulbecco's modified Eagle's medium (without L-glutamine) with 10% fetal bovine serum. The cell density was maintained at a confluence of 70% or less based on a previously described [17]. The cells were treated with 5 mM glutamate (Sigma) or vehicle for 24 hours. The cell viability was investigated by measuring the metabolism of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), as previously described [18]. MTT solution (5 mg/mL) was added to each well of a 96-well plate and incubated for 4 hour at 37℃. After removal of the MTT solution, a solubilization solution containing 50% dimethylformamide and 20% SDS (pH 4.8) was added and incubated overnight. The absorbance was then measured at a wavelength of 570 nm using a microplate reader. The cell viability was represented as the percentage of neuroprotection vs. control set at 100%.
Data analysis
All experimental data are expressed as mean±S.E.M. The intensity analysis was carried out using Sigma Gel 1.0 (Jandel Scientific, San Rafael, CA, USA) and Sigma Plot 4.0 (SPSS Inc., Point Richmond, CA, USA) programs. The ratio of intensity is described as spots intensity of MCAO-operated animal to spots intensity of sham-operated animal. The results in each group were compared by one-way analysis of variance (ANOVA) followed by Student's t-test. The difference for comparison was considered significant at *P<0.05.
Results
We confirmed that the MCAO operation led to brain damage and neuronal cell death using TTC staining (Figure 1). The infarct regions were 2.2±0.5 and 24.8±4.18% in sham-operated and MCAO-induced animals, respectively. Figure 1C shows the histopathological changes in the ischemic region of MCAO operated animals. Neuron in MCAO-induced animals typically had a scalloped and shrunken morphology and apoptotic bodies containing intensive dark masses were found in cell nuclei. However, sham-operated animals had intact neurons in the cerebral cortex. Figure 2 shows change in DLP-1 expression in the cerebral cortices of sham-operated and MCAO-induced animals. The peptide mass of DLP-1 is 14/90 and the sequence of this protein is 21%. DLP-1 protein levels were decreased in MCAO-induced animals compared to sham-operated animals. Western blot analysis clearly indicated a decrease in DLP-1 expression (Figure 3). The levels of DLP-1 were 1.19±0.04 and 0.81±0.03 in the cerebral cortices of sham-operated and MCAO-induced animals, respectively. In the in vitro study, glutamate exposure led to neuronal cell death in cultured HT22 cells (Figure 4A). Moreover, Western blot analysis showed that glutamate toxicity induces decrease in DLP-1 levels in HT22 cells. The levels of DLP-1 were 0.9 3±0.03 and 0.37±0.03 in vehicle-treated and glutamate-treated groups, respectively (Figure 4B, 4C).
Discussion
Cerebral ischemia caused by surgical induction of MCAO induces neuronal cell death in cerebral cortex. The results of this study confirmed that MCAO-induced injury increases infarct regions and apoptotic cell death in the cerebral cortex. In this study, we focused on DLP-1 expression in the cerebral cortex after MCAO and as well as in glutamate-exposed hippocampal neurons. A decrease in DLP-1 protein expression was identified after MCAO-induced brain injury using a proteomic approach. Moreover, Western blot analysis clearly demonstrated that focal cerebral ischemia results in a significant decrease in DLP-1 protein expression.
Oxidative stress is involved in the development of neurodegenerative diseases including stroke, Parkinson's disease, and Alzheimer's disease. Oxidative stress leads to the generation of ROS, ATP depletion, mitochondrial permeability transition pore opening, and results in apoptotic cell death [19]. Mitochondria, a major source of ROS, are involved in the integration of cellular responses to stress. The balance between mitochondrial fusion and fission contributes to the regulation of mitochondrial morphology. Moreover, DLP-1 mediates outer mitochondrial membrane fission, modulates mitochondrial morphology, and regulates mitochondria-dependent cell death [2021]. This reduction in DLP-1 leads to neuronal cell death following ischemic brain injury. DLP-1 depletion induces not only serious brain hypoplasia but also death during embryonic development and after birth in DLP-1 knockout mice [1213]. Decreases in DLP-1 indicate the neuronal disorders. The present study demonstrated that MCAO injury significantly decreases DLP-1 protein levels. Changes in DLP-1 expression during MCAO injury induce mitochondrial dysfunction, resulting in neuronal cell death.
In cultured hippocampal cells, glutamate neurotoxicity generates ROS, increases mitochondrial damage, and consequently leads to cell death [17]. The increase in intracellular Ca2+ by ischemic brain injury induces mitochondrial dysfunction and leads to neuronal cell death via mitochondrial permeability transition [22223]. Mitochondrial dysfunction is a critical step in the pathogenesis of neurodegeneration. Neurodegenerative diseases lead to abnormal mitochondrial dynamics and mitochondrial disorder. This study demonstrated that glutamate treatment leads to ischemic conditions and results in neuronal cell death. Moreover, glutamate exposure significantly decreases DLP-1 levels in HT22 cells. Ischemic conditions cause a dramatic rise in free radical generation that causes cell death [24]. DLP-1 is an important mitochondrial fission and fusion protein that plays an important role in mitochondrial dynamic actions [67]. Imbalances in mitochondrial fission and fusion lead to structural impairment and dysfunction of mitochondria, consequently disrupting ATP generation and causing neuronal cell death [8910]. Decreases in DLP-1 indicate the neuronal damage and dysfunction following glutamate exposure. This study revealed decreased DLP-1 levels in rats with MCAO-induced injury and in glutamate-exposed HT22 cells. These results suggest that decreased DLP-1 expression leads to the imbalance of mitochondrial fission and fusion, resulting in neuronal cell death.

 XML Download
XML Download