

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Alzheimer's disease (AD), which is the most common form of dementia, is characterized by memory loss for recent events, inability to converse, loss of language ability, and affective/personality disturbance and death from opportunistic infections. AD occurs in approximately 10 and 40% of 65 and 80 year olds, respectively. In 2006, there were 26.6 million people worldwide with AD, and it is predicted to affect 1 in 85 individuals globally by 2050 [1]. Brain regions involved in learning and memory processes, including the temporal and frontal lobes, are reduced in size in AD patients owing to degeneration of synapses and death of neurons [2]. Plaques consisting of extracellular deposits of fibrils and amorphous aggregates of Aβ are formed, and diffuse deposits of Aβ are also present in high amounts. Neurofibrillary tangles consisting of intracellular fibrillar aggregates of the microtubule-associated protein, tau, that exhibit hyperphosphorylation and oxidative modifications are also generated [2].
Several hypotheses have been proposed to explain the pathophysiology of AD, including accumulation of Aβ in the brain, disruption of calcium homeostasis, energetic failure, induction of oxidative stress and hyperphosphorylation of the tau protein [34567]. Although a great deal of evidence verifying these hypotheses has been generated, the mechanisms underlying AD and the events responsible for its progression have not been clearly identified.
In the last decade, several approaches for reducing the deposition of Aβ in brain have been studied in basic research and clinical trials, including humanized anti-Aβ monoclonal antibody (bapineuzumab) and γ-secretase inhibitor (LY450139) [4568]. However, this immunotherapy did not show cognitive improvement in a phase 2 trial, despite reducing plasma and cerebrospinal fluid Aβ levels [48]. This is because neuritic atrophy and loss of synapses trigger the pathogenesis of AD, and their dysfunction is a direct cause of memory deficit in AD [4].
Several trials have recently investigated the use of neurosteroids in rebuilding neuronal networks in the damaged brain because neurons with neuritic atrophy may survive and have the potential to be remodeled [49]. Neurosteroids are steroids made by brain cells independently of peripheral steroidogenic sources [10]. These compounds are involved in many neurological functions, including cognitive processes, neurogenesis, mood regulation and addiction [11]. Among these, diosgenin (DG) is a plant-derived steroidal sapogenin of a major constituent in the Dioscorea rhizome and other herbal drugs, such as those from Trigonella spp., Polygonatum spp. and Smilax spp. [9]. DG also serves as an important starting material for the production of corticosteroids, sexual hormones, oral contraceptives as well as other steroidal drugs via hemisynthesis [12]. Furthermore, DG has several biological effects, such as anti-cancer [13], anti-food allergy [14] and anti-cognitive deficit [15] activity and the ability to relieve diabetic neuropathy [16]. A few recent studies have also shown that DG induces cognitive enhancement and results in memory recovery in 5XFAD mice coexpressing the amyloid beta precursor protein (APP) and presenilin 1 (PS1) mutant gene [917]. However, the therapeutic effects of DG as a nerve growth factor (NGF) stimulator on multiple types of brain damage have never been investigated using an animal model of Aβ peptide accumulation and neurotoxicant-induced cell death.
The present study was conducted to investigate the beneficial effects of DG on Aβ accumulation and neuronal cell death through the regulation of NGF biosynthesis in transgenic 2576 (TG) mice with multiple types of neuronal damage induced via Aβ-42 accumulation and trimethyltin (TMT) injection.
Materials and Methods
Care and use of animals and experimental design
The animal protocols used in this study were reviewed and approved based on the ethical procedures and scientific care of animals set by the Pusan National University-Institutional Animal Care and Use Committee (PNU-IACUC; Approval Number PNU-2014-0628). Adult TG mice and non-transgenic (nTG) mice (B6SJLF1/J, 20±5 g) were purchased from Samtaco-Bio Korea (Osan, Korea) and raised to be an average of 15 months old at the Pusan National University Laboratory Animal Resources Center, which is accredited by AAALAC International (Accredited Unit Number-001525) and the Korea Food and Drug Administration (KFDA; Accredited Unit Number-000231). All mice were given a standard irradiated chow diet (Purina Mills, Seoungnam, Korea) ad libitum, and were maintained in a specific pathogen-free state under a strict light cycle (lights on at 08:00 h and off at 20:00 h) at 23±2℃ and 50±10% relative humidity.
Mice an average age of 15 months (n=28) were first assigned into two groups: nTG mice (nTG group; n=7) and TG mice (TG group; n=21). The TG group was further divided to one of the following three groups, a vehicle (VC) treated group (TG+VC group, n=7), DG treated group (TG+DG group, n=7) and memantine hydrochloride (MT) treated group (TG+MT group, n=7). The TG+VC group received a comparable volume of olive oil (Sigma-Aldrich Co., St. Louis, MO, USA) per body weight, whereas the TG+DG group received 10 µM/kg/body weight of DG (Sigma-Aldrich Co.), and the TG+MT group received 10 mg/kg/body weight of MT (Tocris Bioscience, Tocris House, Bristol, UK). Treatments were administered intraperitoneally (i.p.) to mice twice a day for 21 days. At 21 days, all mice in the TG group were injected i.p. with a single dose of TMT (Sigma-Aldrich Co.) (2.5 mg TMT/kg body weight) dissolved in 1× PBS. After 3 days, all mice were sacrificed using Zoletile (150 mg/kg body weight) to acquire blood and tissue samples for further analysis.
Histological analysis
Brain perfusion and Nissl staining were performed as previously described [18]. Briefly, mice were anaesthetized by intraperitoneal injection of Zoletile (150 mg/kg body weight), then transcardially perfused with 1× PBS followed by neutral buffered formaldehyde to effectively remove blood and fix brain tissue. Following perfusion, each mouse brain was isolated from the skull and fixed overnight in neutral buffered formaldehyde, after which each brain was dehydrated and embedded in paraffin. Next, a series of brain sections (10 µm) were cut from paraffin-embedded tissue using a Leica microtome (Leica Microsystems, Bannockburn, IL, USA). For Nissl staining, these sections were deparaffinized with xylene, rehydrated with ethanol at graded decreasing concentrations of 100–70%, and finally washed with distilled water (dH2O). The slides with brain sections were subjected to Nissl staining solution with 0.1% cresyl violet acetate for 8 min, then washed with dH2O, after which the dead neuronal cells were enumerated using Leica Application Suite (Leica Microsystems, Wetzlar, Germany).
For immunohistochemical analysis for Aβ-42 peptides, the brain sections were deparaffinized with xylene, rehydrated, and pretreated for 30 min at room temperature with PBS blocking buffer containing 10% normal goat serum (Vector Laboratories Inc. Burlingame, CA, USA). Next, the sections were incubated with mouse anti-Aβ-42 peptide antibody (Chemicon International, Inc. Billerica, MA, USA) at a dilution of 1:100 in tris-buffered saline (TBS) blocking buffer for 12 h. The antigen-antibody complexes were visualized with biotinylated secondary antibody (goat anti-mouse)-conjugated HRP streptavidin (Histostain-Plus Kit; Zymed, South San Francisco, CA, USA) at a dilution of 1:100 in TBS blocking buffer. Aβ peptides were detected using stable 3,3'-diaminobenzidine (DAB; Invitrogen, Carlsbad, CA, USA) and observed with Leica Application Suite (Leica Microsystems).
Slot blot analysis
Brain tissue including the cortex tissue (45 mg) and the hippocampus tissue (5 mg) was chopped with surgical scissors and homogenized with a glass homogenizer in PRO-PREP protein extraction solution (iNtRON Biotechnology Inc., Seongnam, Korea) containing 1.0 mM phenylmethylsulfonyl fluoride (PMSF), 1.0 mM ethylenediamine tetraacetic acid (EDTA), 1 µM pepstatin A, 1 µM leupeptin, and 1 µM aprotinin. The resulting mixture was then centrifuged for 10 min at 13,000 rpm at 4℃. The concentrations of the separated proteins were determined using a BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA), and 12.5 µg protein were transferred to a nitrocellulose membrane using a Slot Blot kit (Amersham Pharmacia Biotech, Piscataway, NJ, USA). The membrane was incubated separately with primary rabbit anti-Aβ-42 peptide (Invitrogen, Carlsbad, CA, USA) at 2 µg and mouse anti-Aβ peptide antibody (Chemicon International Inc.) at a dilution of 1:1,000 in blocking buffer at room temperature for 1 h, then washed with washing buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 0.05% Tween 20), then incubated with horseradish peroxidase (HRP)-conjugated goat antirabbit IgG (Invitrogen) and HRP-conjugated goat antimouse IgG (Invitrogen) at a 1:1,000 dilution and room temperature for 1 h. Membrane blots were developed using Amersham ECL Select Western Blotting detection reagent (GE Healthcare, Little Chalfont, UK). The signal image from each protein was acquired by a digital camera (1.92 MP resolution) in conjunction with the FluorChem® FC2 Imaging System (Alpha Innotech Co., San Leandro, CA, USA), and their density was quantitated by scanning with the AlphaView Program V3.2.2 (Cell Bioscience Inc., Santa Clara, CA, USA). Total proteins of three samples from each group were analyzed in two separate western blot analyses.
Determination of acetylcholinesterase (AChE) activity
AChE assay was performed using an Acetylcholinesterase Assay Kit (Abcam, Cambridge, UK) according to the manufacturer's protocols. Briefly, the brain tissue was homogenized in 0.1M phosphate buffer (Sigma-Aldrich Co.), after which the homogenates were stored at -70℃ until analysis. The sample or standards and AChE reaction mixture were then incubated on a 96-well plate for 20 min at room temperature while protected from the light. Color alterations were read using a Versa max plate reader (Molecular Devices, Sunnyvale, CA, USA) at 405 nm.
ELISA for NGF
The levels of NGF in the brain tissue were measured using a NGF ELISA kit (Chemicon International Inc.) according to the manufacturer's protocols. The brain tissue for ELISA was prepared by homogenization in NGF lysis buffer as previously described [16]. Samples and standards were incubated overnight on antibodycoated plates in a plate shaker at 100–150 rpm and 2–8℃. The wells were then washed four times with washing buffer, after which 100 µL of anti-mouse NGF antibody was added to each well. Plates were subsequently incubated in a shaker for 2 h at room temperature, after which 100 µL of peroxidase-conjugated donkey anti-mouse IgG polyclonal antibody was added to each well and the samples were incubated at room temperature for 2 h. After washing, 100 µL of TMB/E substrate was added to each well and the plate was incubated at room temperature for 15 min. The reaction was subsequently quenched by the addition of 100 mL stop solution, after which the plates were analyzed by evaluation of the absorbance at 450 nm using a Versa max plate reader (Molecular Devices, Sunnyvale, CA, USA).
Western blot
Total proteins prepared from the brain tissue were separated by 4–20% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) for 2 h, after which resolved proteins were transferred to nitrocellulose membranes for 2 h at 40 V. Each membrane was then incubated separately at 4℃ with the following primary antibodies overnight: anti-NGF (Abcam), anti-TrkA (Cell Signaling, Danvers, MA, USA), anti-p-TrkA (Cell Signaling), anti-Akt (Cell Signaling), anti-p-Akt (Cell Signaling), anti-ERK (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-p-ERK (Santa Cruz Biotechnology), anti-p-75NTR (Abcam), anti-JNK (Cell Signaling), anti-p-JNK (Cell Signaling), anti-Bcl2 (Abcam), anti-Bax (Abcam), and anti-β-actin antibody (Sigma-Aldrich Co.). The membranes were then washed with washing buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 0.05% Tween 20) and incubated with HRP-conjugated goat anti-rabbit IgG (Invitrogen) and HRP-conjugated goat anti-mouse IgG (Invitrogen) at a 1:1,000 dilution and room temperature for 1 h. Membrane blots were developed using Amersham ECL Select Western Blotting detection reagent (GE Healthcare).
Determination of malondialdehyde (MDA) levels
The MDA levels in the brain sample were assayed using a Lipid Peroxidation (MDA) Assay Kit (Sigma-Aldrich Co.) according to the manufacturer's protocols. Briefly, 45 mg cortex and 5 mg hippocampus tissue from each group of mice were homogenized in MDA lysis buffer containing butylhydroxytoluene (BHT), after which the homogenates were stored at -20℃ until analysis. The sample or standards and TBA solution (70 mM thiobarbituric acid and 5M glacial acetic acid) were incubated at 95℃ for 60 min, then cooled to room temperature in an ice bath for 10 min, after which the reaction absorbance at 532 nm was read using a Versa max plate reader (Molecular Devices).
Analysis of superoxide dismutase (SOD) activity
SOD activity in the brain tissue was detected using a calorimetric assay and the reagents in a SOD Assay Kit (Dojindo Molecular Technologies Inc., Japan). First, the brain tissues of mice from each group were homogenized in 500 µL of sucrose buffer (0.25 mol/L sucrose, 10 mmol/L HEPES, 1 mmol/L EDTA, pH 7.4). The lysate was then harvested from the mixture by centrifugation at 10,000×g for 60 min and diluted with dilution buffer or saline as follows: 1, 1/5, 1/52, 1/53, 1/54, 1/55, 1/56. Next, 25 mL aliquots of each sample solution were placed in 96 well plates, after which 200 mL of the WST working solution was added. In addition, an enzyme working solution (20 µL) was added to each well and the samples were mixed thoroughly. The enzyme reaction was induced by incubating the mixture plate at 37℃ for 20 min, after which the absorbance at 450 nm was measured using a spectrophotometer. The SOD activity was calculated directly using the following equation: SOD activity (inhibition rate %)=[(Ablank 1-Ablank 3)-(Asample-Ablank 2)]/(Ablank 1-Ablank 3)×100 (Ablank 1: absorbance of blank 1, Ablank 2: absorbance of blank 2, Ablank 3: absorbance of blank 3, Asample: absorbance of sample).
Statistical analysis
One-way ANOVA was used to identify significant differences between nTG and TG mice (SPSS for Windows, Release 10.10, Standard Version, Chicago, IL, USA). Additionally, differences between the TG+VC group and the TG+DG or TG+MT group were evaluated by a post hoc test (SPSS for Windows, Release 10.10, Standard Version) of the variance and significance levels. All values were expressed as the means ± SD. A P value of <0.05 was considered significant.
Results
Induction of multiple types of brain damage in TG mice
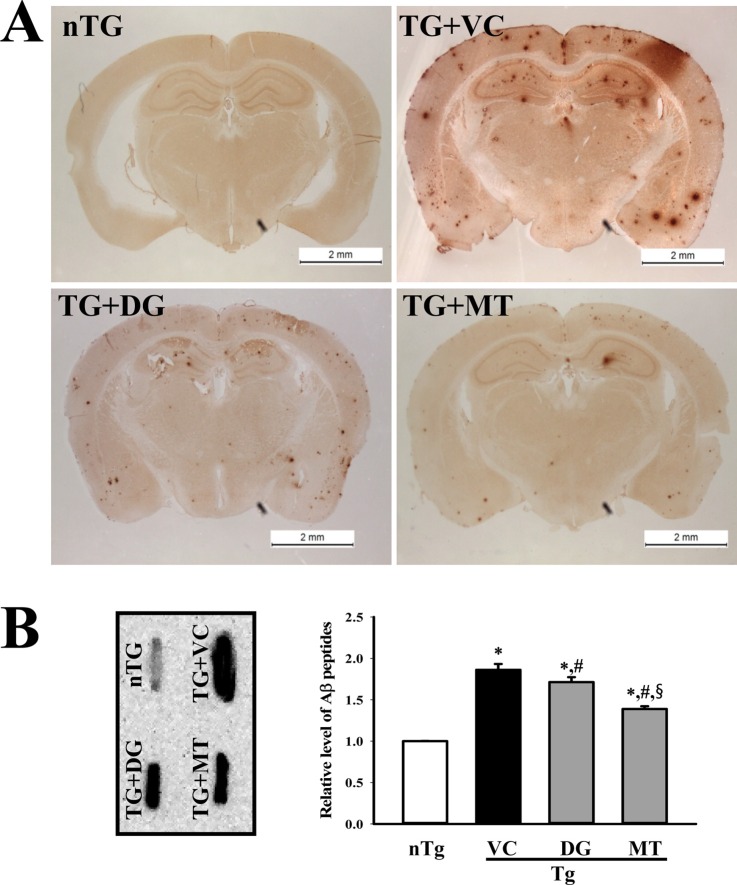
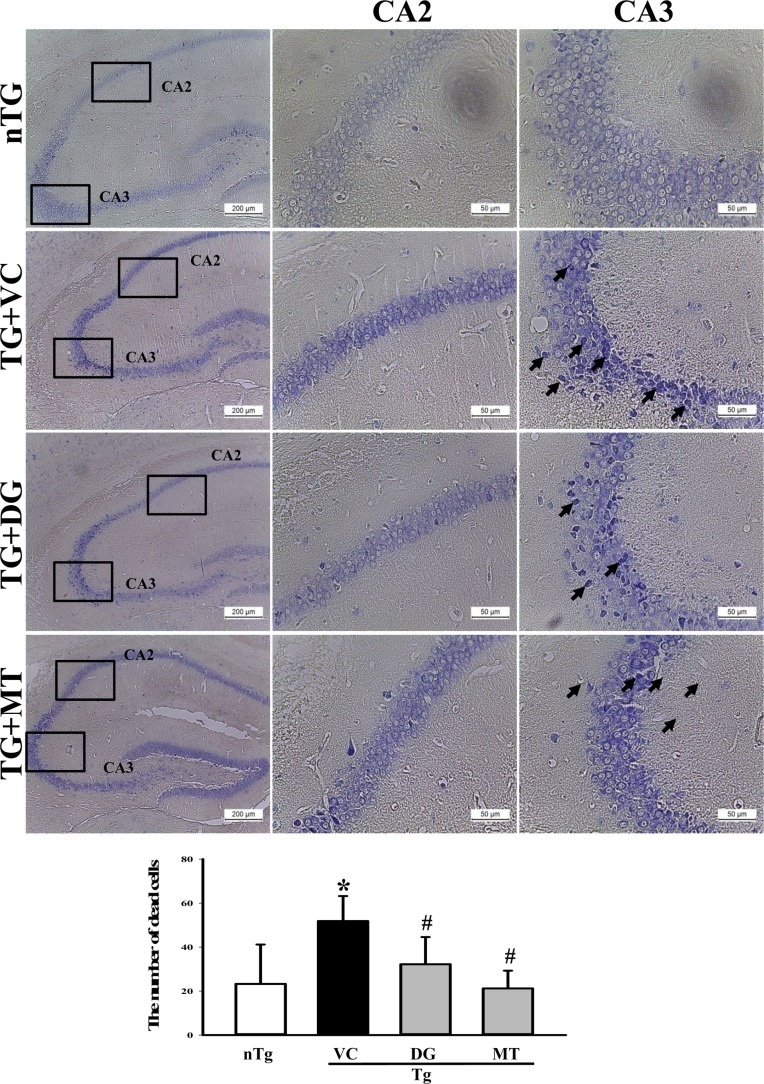
To induce multiple types of brain damage, including Aβ-42 accumulation and neuronal cell death, TMT was injected into TG mice overexpressing APPsw proteins. The number of Aβ-42 stained plaques and Nissl stained neuronal cells increased significantly in TMT-injected TG mice (TG+VC group) compared with nTG mice (Figure 1A and 2A). These results indicate that multiple types of brain damage associated with high levels of Aβ-42 peptides and neuronal cell death can be successfully induced by TMT injection in TG mice.
Effect of DG on Aβ accumulation in TMT treated TG mice
To investigate the beneficial effects of DG on Aβ accumulation in model mice with multiple types of brain damage, the Aβ accumulation and concentration were measured in the TG+DG group after DG pretreatment for 21 days. Generally, TG mice overexpressing the Swedish mutant form (KM670/671NL) of APP (isoform 695) developed numerous parenchymal Aβ plaques by 11–13 months with some vascular [19]. As shown in Figure 1, a number of Aβ-positive stains were shown in the cerebral cortex and hippocampus of the TG+VC group, while Aβ plaques were not observed in the agematched nTG mice. However, the TG+DG mice displayed a significant decrease in Aβ in the cerebral cortex and hippocampus (Figure 1A). Aβ plaques were also significantly reduced in the cortex and hippocampus of TG+MT group mice (Figure 1A). Our dot blot data are also consistent with the immunohistochemistry results. Total Aβ peptides in the cortex and hippocampus tissue were most abundant in TG+VC group, but significantly reduced after DG or MT treatment (Figure 1B). Taken together, these results indicate that DG treatment can reduce Aβ accumulation and production in response to multiple types of brain damage.
Effect of DG on neuronal cell death in TMT treated TG mice
To examine the protective role of DG on neuronal cell death, alterations in the number of dead cells were measured in the brain of TMT treated TG mice after Nissl staining analysis. This analysis can specifically stain the Nissl body, which is a large granular body found in neurons, and is used to identify neuronal apoptosis. As shown in Figure 2, dead cells reported to be dark blue were abundant in the hippocampus CA2 and CA3 region of the TG+VC group compared with the nTG group. However, the number of these cells was significantly lower in some regions of the TG+DG and TG+MT group (Figure 2). These results indicate that DG may greatly decrease the number of TMT induced dead cells in the CA2 and CA3 hippocampus of the multiple brain disease model.
Effect of DG on cerebral cholinergic function and cell death
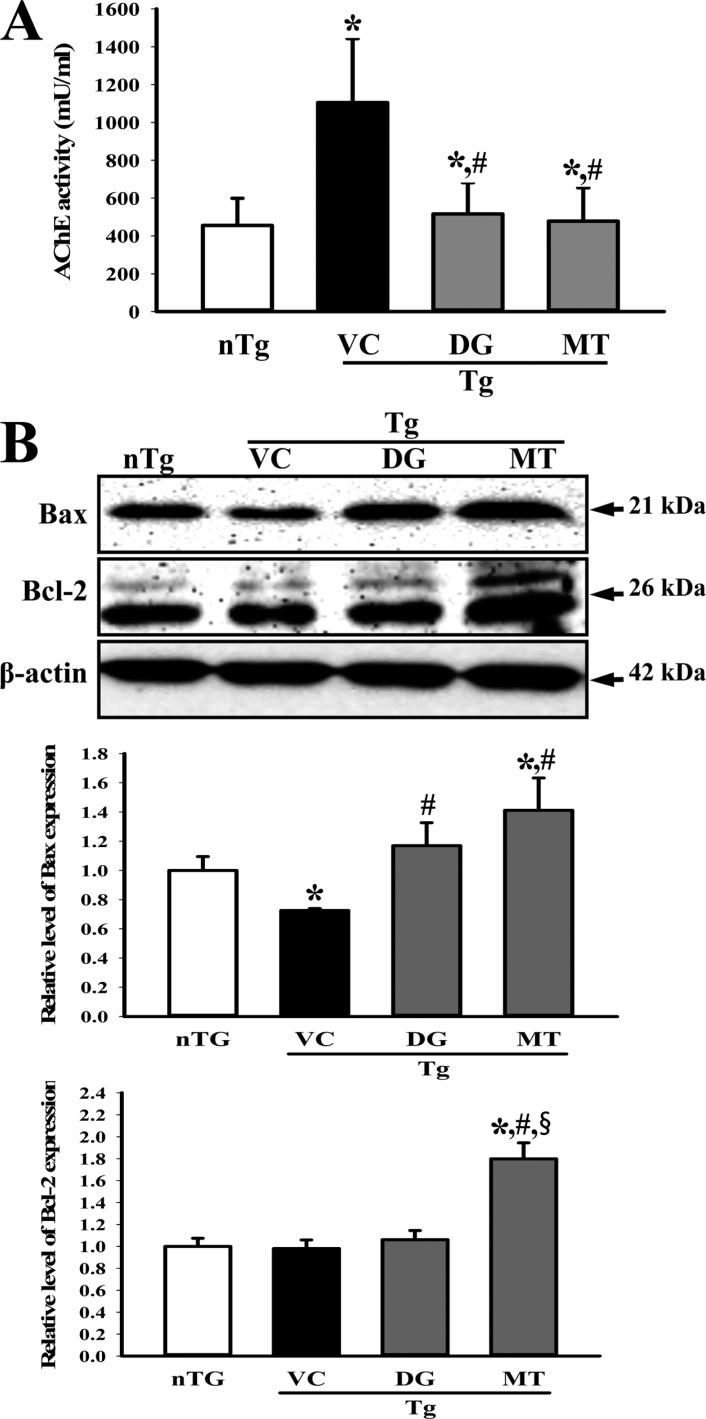
AChE is an enzyme that catalyzes the breakdown of acetylcholine and some other choline esters that function as neurotransmitters. AChE is mainly found at neuromuscular junctions and in chemical synapses of the cholinergic type, where its activity serves to terminate synaptic transmission [20]. To determine the effects of DG on cerebral cholinergic function, the activities of AChE were evaluated in the cerebral cortex and hippocampus of multiple brain disease model mice pretreated with DG. As shown in Figure 3A, AChE activity in the TG group exposed to VC solution was highly upregulated when compared with the age-matched nTG group. However, the AChE activity was rapidly recovered in the TG+DG or TG+MT group (Figure 3A). Therefore, the above results indicate that DG treatment is tightly correlated with regulation of AChE activity.
The expression of apoptosis related proteins was observed in subset groups to examine the protective effects of DG. Significant recovery was detected in the TG+DG group or TG+MT group when compared with the nTG group (Figure 3B).
Effect of DG on NGF biosynthesis and NGF receptor signaling pathway
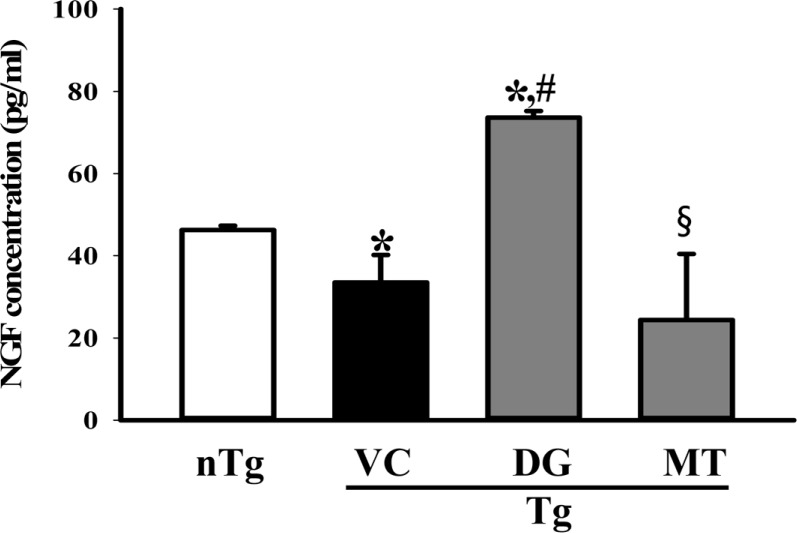
To investigate the role of DG as a NGF stimulator under the mechanism involved in DG-mediated neuroprotection, alterations in NGF biosynthesis and the NGF signaling pathway were investigated in the brains of the TG+DG group. First, the NGF concentrations in the brain tissues of the subset group were measured using an anti-NGF ELISA kit. The level of NGF in the lysates from the cortex and hippocampus tissue was lower in the TG+VC group than the nTG group. However, these levels were dramatically increased with 124% in DG pretreated TG mice, although the TG+MT group was maintained at a constant level (Figure 4).
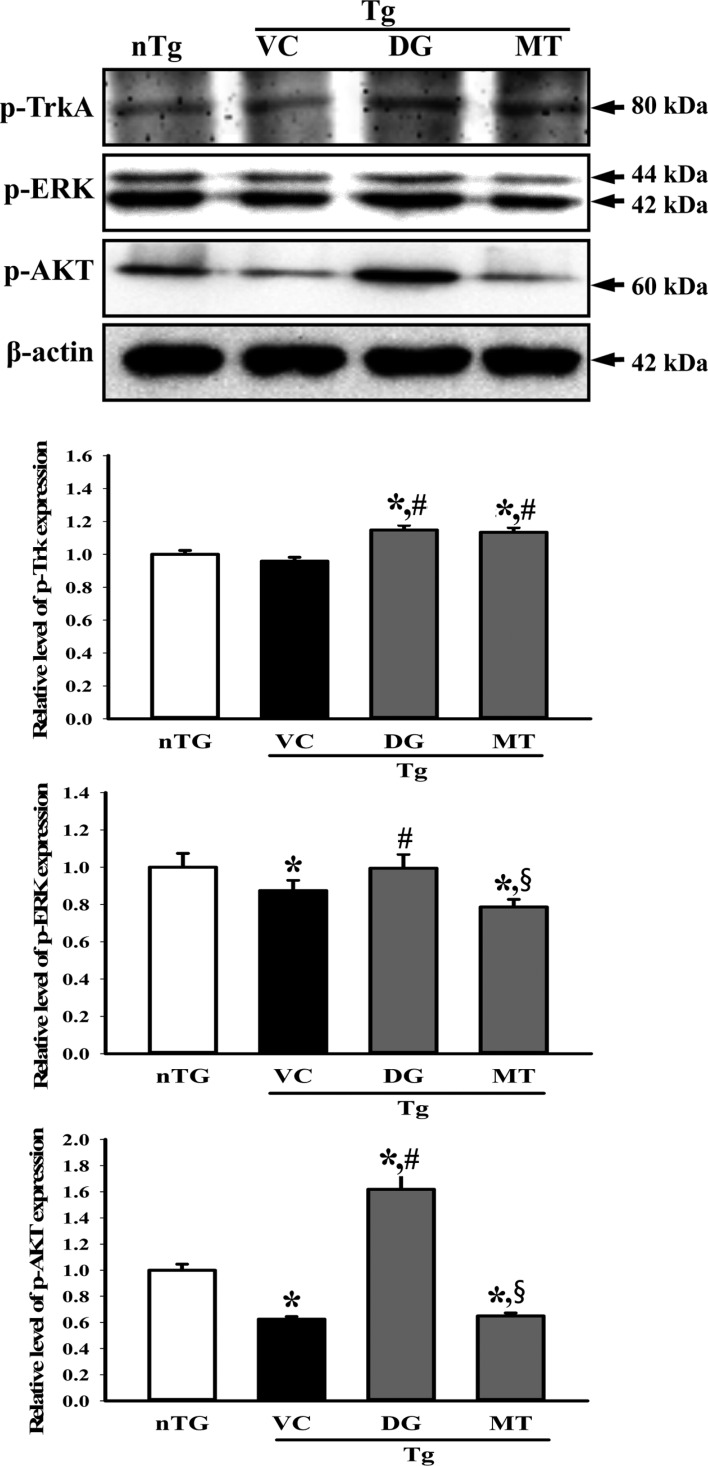
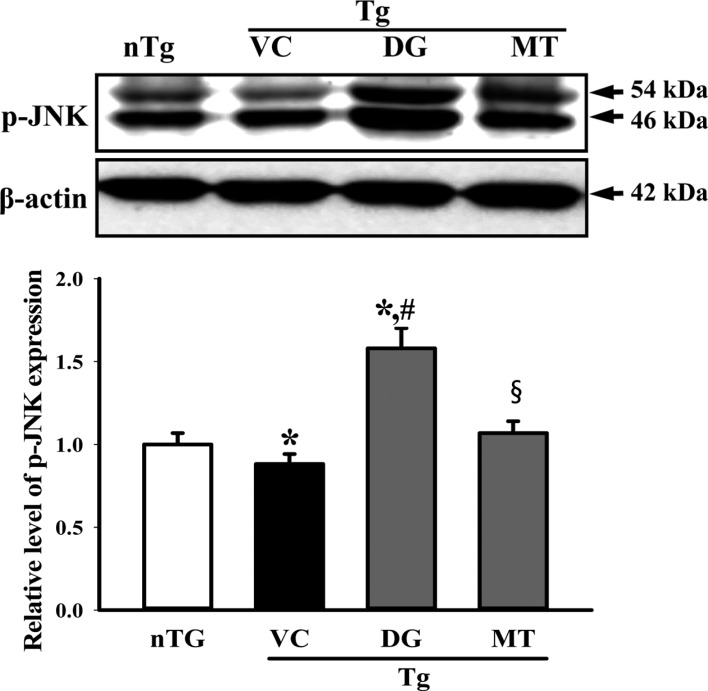
Since DG induces NGF secretion in the brains of multiple brain damage models, we assumed that DG can stimulate the NGF signaling pathway. To further examine whether DG augments the NGF signaling pathway, we measured expressional alteration of the downstream signaling pathway components including TrkA, ERK and Akt in the lysate mixture prepared from the cortex and hippocampus tissue using the specific primary antibody. The level of p-TrkA, p-ERK and p-AKT was lower in the TG+VC group than the nTG group although p-TrkA did not show any significant. These levels were increased in the TG+DG group, but the increase ratio varied (Figure 5). Moreover, a similar enhancement was observed in the p-JNK level of the TG+DG brain. DG pretreatment significantly increased with 98.7% the expression level of p-JNK (Figure 6).
Overall, these results show that DG as a stimulator of NGF can induce NGF biosynthesis. Furthermore, NGF induced by DG treatment may regulate the survival, proliferation and apoptosis of neuronal cells through the NGF receptor signaling pathway.
Antioxidant effect of DG
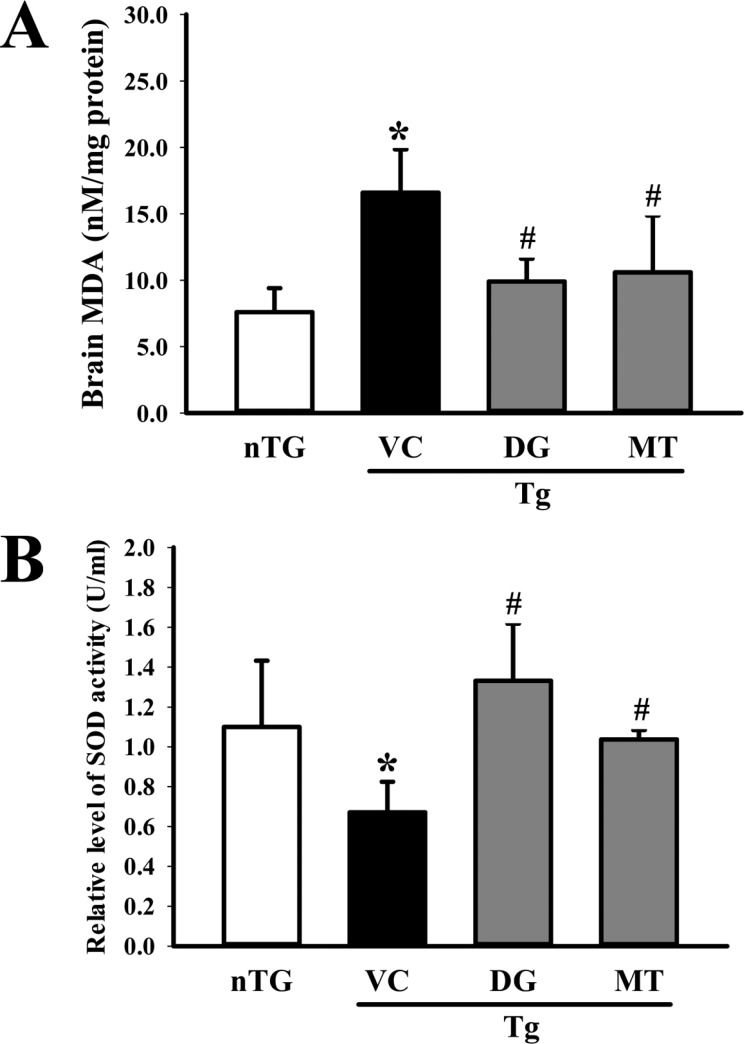
SOD can scavenge superoxide radicals (O2-), which can lead to fatal damage to cells; therefore, it plays a crucial role in the defense against oxidative damage. To investigate whether DG activity in the neuronal cells was correlated with anti-oxidant properties, alterations in SOD activity were measured in the homogenate of the cortex and hippocampus tissue collected from subset groups. SOD activity was higher with 85.7% in TG+DG group than the TG+VC or age-matched nTG group (Figure 7B). The opposite pattern was observed in the MDA concentration. The TG+VC group showed a higher level of MDA than the nTG group, although this level was significantly decreased in the TG+DG group (Figure 7A). Therefore, these results indicate that DG can contribute to the up-regulation of anti-oxidant activity in brains exposed to multiple types of damage.
Discussion
NGF is a potential biological activator that prevents neuronal cell death, promoting neuritic outgrowth and differentiation, and maintaining neuron organization [21]. It is expected to be applied to treatment of neurodegenerative diseases including AD, Parkinson's disease and amyotrophic lateral sclerosis [2223]. Therefore, identification of novel NGF stimulator has been considered an important strategy for treatment of neurodegenerative disease because NGF is unable to cross the blood-brain barrier [24]. Several studies have reported low molecular weight compounds that stimulate NGF synthesis such as catecholamins [25], scabronions [26], cymeines [27], hericenones and erinacines [28]. In this study, the neuroprotective effects of DG were investigated to determine if DG pretreatment could stimulate NGF biosynthesis and secretion in the brain following multiple types of damage including Aβ-42 deposition and TMT-induced neuronal death. The results presented herein are the first to demonstrate that DG as a NGF stimulator will likely impact the development and application of protection drugs.
Among various neuronal diseases, AD is a well known neurodegenerative disorder characterized by gradual loss of cognitive ability [29]. The main pathological features of AD are neuronal loss, the deposition of senile Aβ protein plaques and intracellular neurofibrillary tangles in some brain regions [30]. Aβ is one of the main factors that leads to the progression of AD. Aβ is highly toxic to neurons and can trigger a cascade of pathogenic processes that lead to cell death [3132]. Treatment with DG significantly reduced amyloid plaques and neurofibrillary tangles in the cerebral cortex and hippocampus of 5XFAD mice, although the correlation between their function and NGF stimulators was not investigated [917]. Similar results in Aβ deposition were detected in the present study. Our immunohistochemistry results showed that the TG+DG group containing multiple types of brain damage exhibited a significant decrease of Aβ in the cerebral cortex and hippocampus as shown Figure 1.
In the AD brain, the degree of amnesia and cerebral deposition of Aβ is correlated with marked cholinergic dysfunction. AD causes cholinergic deficiency owing to the degeneration or atrophy of cholinergic neurons in the brain. AChE is a neurotransmitter, and its activity serves to terminate synaptic transmission. Several reports have shown that AChE in AD patients causes different degrees of damage [333435]. The decrease of neurotransmitter ACh in the brain has been shown to be followed by learning and memory deficits [36]. AChE are AChspecific hydrolases, and augmented AChE activity may lead to reduced ACh levels. Several studies have proposed that AChE inhibitors such as rivastigmine, galantamine, and aricept increase the level of ACh and effectively improve the cognitive function [373839]. Our results indicate that TG2576 AD model mice showing Aβ accumulation have high AChE activity, but that DG pretreatment induced a significant decrease in AChE activity, indicating that DG can be a potent therapeutic solution for AD rescue.
Our results also indicate that DG stimulates the biosynthesis of NGF and activates downstream molecules such as p-TrkA, p-ERK, p-AKT, and p-JNK in NGF signaling (Figures 5 and 6). Several compounds and extracts have been reported to be NGF stimulators. Propentofylline, a xanthine derivative, stimulated the synthesis and secretion of NGF from mouse astrocytes, while hericenones and erinacines from the fruiting body and mycelium of Hericium erinaceus induced NGF biosynthesis in mouse astroglial cells [2840]. DG from Dioscorea nipponica increased the NGF level in the sciatic nerve of diabetic polyneuropathy [16]. In the present study, we used TMT-injected TG2576 mice as a multiple brain damage model containing Aβ-42 accumulation and nerutoxicant-induced death. DG successfully induced the biosynthesis and secretion of NGF in this model. The results presented herein are the first to provide additional evidence that DG can act as a novel compound to treat neurodegenerative disorders, although more studies are needed to determine the optimal concentration and toxicity.
Taken together, DG can act as a neuroprotectant against multiple types of brain damage with Aβ accumulation and TMT-induced neuronal death. First, DG inhibits neural cell death by reducing Aβ accumulation, upregulating SOD activity and suppressing lipid peroxidation. DG then recovers cerebral cholinergic function by enhancing AChE activity. Finally, DG blocks neural cell death by accelerating NGF expression and stimulating the NGF receptor signaling pathway. Accordingly, our results suggest that administration of DG may provide a therapeutic approach for AD.
 XML Download
XML Download