

 PDF
PDF ePub
ePub Citation
Citation Print
Print


The human pathogen Streptococcus (S) pneumoniae is known to cause a variety of diseases, such as community-acquired pneumonia, meningitis, septicemia, otitis media and sinusitis [1]. Pneumococcal pneumonia is especially common among children and elderly people, who can have weak immune systems. Despite antibiotic therapy, S. pneumoniae continues to induce considerable morbidity and mortality worldwide. Approximately 800,000 children younger than 5 years die because of S. pneumoniae infections worldwide each year, with 90% of cases occurring in developing countries [2]. Despite its importance as a major human pathogen, interactions between the host and S. pneumoniae are not clearly understood. Therefore, investigations into the host immune defense mechanisms against S. pneumoniae in an animal model are required to better understand and control these infections. Tumor necrosis factor-alpha (TNF-α) is a pro-inflammatory cytokine mainly produced by macrophages, and to a lesser extent by B cells, T cells, NK cells, Kupffer cells, glial cells and adipocytes. [3]. It regulates cell proliferation, differentiation, apoptosis, inflammation and the immune response [4]. TNF-α reportedly plays an important role in mice infected with S. pneumoniae. Compared with control mice, mice treated with antibodies against TNF-α or TNF-α receptor-deficient mice showed increased bacterial counts, along with early and high mortality after intranasal S. pneumoniae infection [56]. However, these studies did not determine the cause of early death in the mice. To elucidate the role of TNF-α in S. pneumoniae infections, and to determine the cause of early death in TNF-α deficient mice, we used a murine pneumococcal pneumonia model involving TNF-α knockout (KO) mice.
Materials and Methods
Animals
We used male wild-type (WT) C57BL/6J and TNF-α KO (B6.129S-Tnftm1Gkl/J) mice that were 7-8 weeks old in this study. The WT mice were donated by the Korea Research Institute of Bioscience and Biotechnology (Chungbuk, Korea). The TNF-α KO mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Mice were bred at a specific pathogen-free facility at the College of Veterinary Medicine (Konkuk University). Mice were housed in sterilized polycarbonate cages with sterilized chip bedding and allowed free access to a sterilized pellet diet and water. The room containing the mice had a controlled 12 h light-dark cycle, and the temperature was maintained at 22±2℃ with 50±10% relative humidity. This study was approved by the Institutional Animal Care and Use Committee (IACUC) of Konkuk University (approval No: KU10026).
Preparation of bacteria
We obtained the S. pneumoniae D39 serotype 2 from the Korea Centers for Disease Control and Prevention. D39 is the reference strain of S. pneumoniae, and is the most widely studied strain. It is highly encapsulated and results in a lethal outcome following infection. We cultured S. pneumoniae D39 on 5% sheep blood agar plates (Medexx) at 37℃/5% CO2 for 18 h before harvesting. Harvested S. pneumoniae D39 was rinsed and resuspended in sterilized phosphate-buffered saline (PBS) then transferred to brain heart broth (Merck) and cultured without shaking for 6 h at 37℃/5% CO2. The S. pneumoniae D39 cultures were centrifuged, washed and rinsed with sterile PBS. The infectious dose was determined by counting colony forming units (CFU) after plating serially diluted S. pneumoniae D39 suspensions on 5% sheep blood agar plates. Plates were cultured at 37℃/5% CO2 for 24 h.
Intranasal infection with S. pneumoniae D39
Mice were anesthetized by intraperitoneal injections of Zoletil (40 mg/kg; Virbac Laboratories) and Rompun (5 mg/kg; Bayer Korea). Mice (n=30 WT, n=30 KO) were intranasally infected, using a pipette, with 20 µL of inoculum containing 2×107 CFU of S. pneumoniae D39. The survival rate and body weight of 10 WT and 10 KO mice were monitored at 12, 24, 48 and 72 h post-infection. For the remaining mice, five WT and five KO mice were euthanized at 12, 24, 48, and 72 h post-infection; blood was collected from the caudal vena cava. The trachea, lungs and spleen were removed from mice and kept for other experiments.
Differential cell counts in bronchoalveolar lavage (BAL) fluid
During necropsy, the trachea and lungs were removed, and the trachea cannulated with a sterilized 23-gauge needle. We injected 0.4 mL of PBS into the lungs and then collected BAL fluid; this process was repeated three times. The BAL fluid was centrifuged and the supernatant discarded. The pellet was resuspended in 0.5 mL of PBS and differential cell counts were performed with a FORCYTE hematology analyzer (Oxford Science Inc.).
Bacterial counts in the lungs and blood
We conducted bacterial counts in the lungs and blood. Approximately one-third of the left lobe of the lungs, and blood, were frozen in liquid nitrogen until required. Lung tissue was homogenized by a tissue homogenizer and serially diluted in PBS; blood samples were also serially diluted in PBS. We plated 100 µL of serially diluted lung homogenates or blood samples on 5% sheep blood agar plates and cultured for 24 h at 37℃/5% CO2. The number of colonies was counted and bacterial counts calculated and expressed as a log number of CFUs per 0.1 g of lung homogenate or 1 mL of blood.
Histopathological analysis
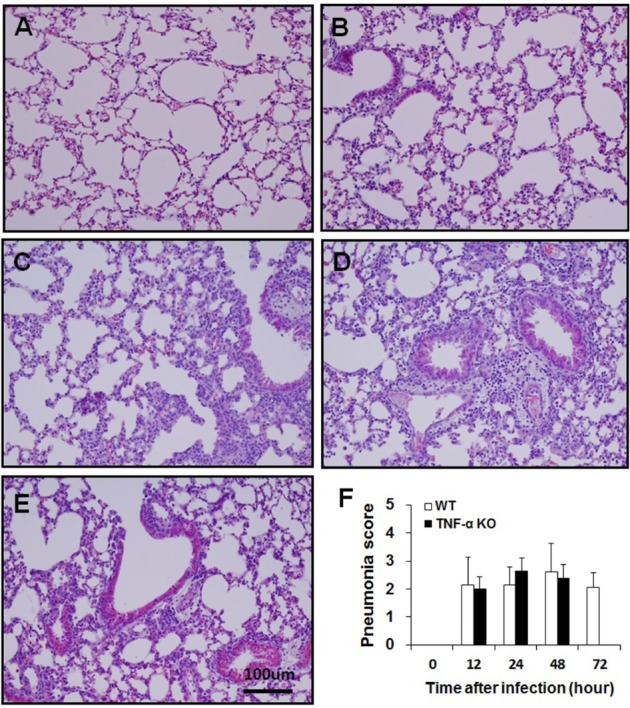
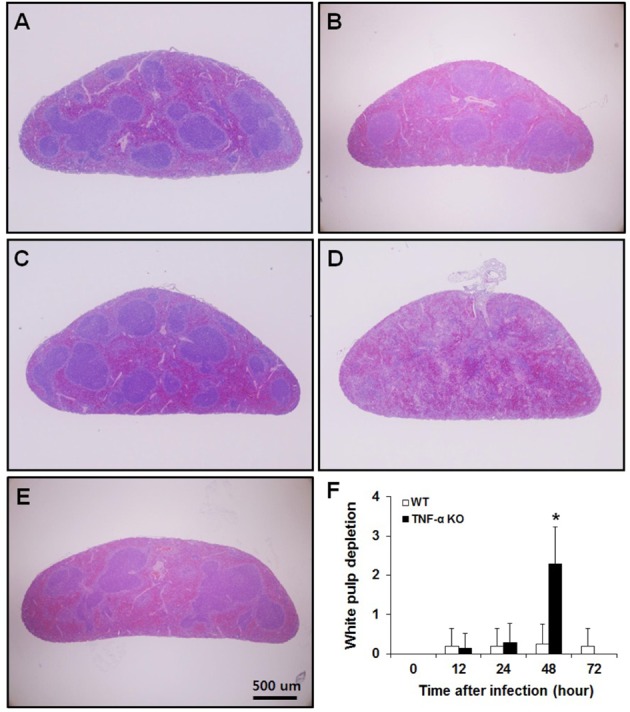
One-third of the left lobe of the lung and one-third of the spleen were fixed in 10% neutral-buffered formalin solution and processed routinely for paraffin-embedding. Sections were cut from paraffin blocks (4-µm thickness) and stained with hematoxylin and eosin (H&E). To quantify the degree of pneumonia, nine fields of view for each lung tissue section at 200× were randomly examined and scored as described in a previous study [7]. Briefly, intra-alveolar inflammation, bronchitis, edema and endothelialitis were scored 0-3 as follows: 0, absent; 1, mild; 2, moderate; and 3, severe. The total pneumonia score was expressed as the sum of the scores for each parameter with a possible maximum score of 12. To compare the histopathological lesions in the spleen, a section of spleen tissue was examined at ×40 and white pulp depletion was scored 0-3 as follows: 0, absent; 1, mild; 2, moderate; and 3, severe.
TUNEL assays
Spleen sections were stained with a commercially available apoptosis detection kit (ApopTag, Chemicon international, Inc.,) according to the manufacturer's instructions. Briefly, the sections, deparaffined and hydrated, were treated with proteinase K (20 µg/mL) in PBS (pH 7.5) at 37℃ for 20 min and then treated 3% hydroperoxidase in PBS to block endogenous peroxidase. After washing with PBS, the slides were incubated with terminal deoxynucleotidyl transferase and a nucleotide mixture at 37℃ for 1 h and then applied for 30 min with anti-digoxigenin conjugate. The slides were treated with DAB, before counterstain with hematoxylin. After dehydration and cleaning, the sections were coverslipped and then examined under a microscope (Olympus BX51). To examine the white pulp, five random fields of view were examined for each spleen section. Apoptotic cells were analyzed with MetaMorph 7.5 (Molecular Devices) image analysis software by an experienced pathologist. Results were expressed as the number of apoptotic cells per 1 mm2.
Cytokine analysis
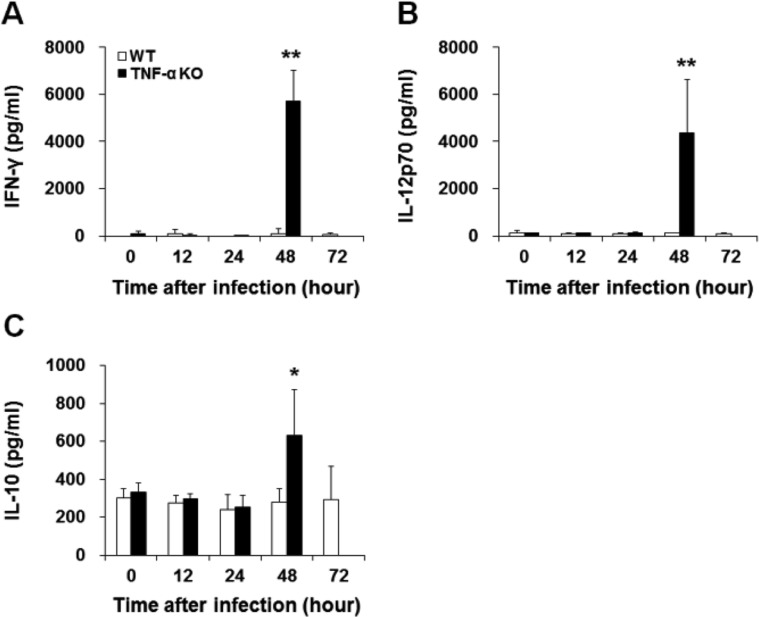
Blood was collected from the caudal vena cava and stored at 4℃ overnight. Samples were centrifuged (12,000 rpm, 10 min), supernatants collected and then frozen at -70℃ until required. The levels of IFN-γ, IL-12p70 and IL-10 in serum were measured using commercially available enzyme-linked immunosorbent assay (ELISA) kits (eBioscience) according to the manufacturer's instructions. Cytokine concentrations were determined from absorbance readings and through the use of graphs for known standards of each cytokine. The detection limits of IFN-γ, IL-12p70 and IL-10 were 15, 15 and 8 pg/mL, respectively.
Statistical analysis
Data are expressed as means±standard deviation (SD). Significant differences between WT and KO mice were evaluated using Student's unpaired t-test. Survival rates were compared using the log-rank test. Data analysis was conducted with GraphPad Prism 5 for Windows (GraphPad Software). P-values less than 0.05 were considered statistically significant.
Results
Survival rate and body weight changes
Following infection, appearance, behavior, body weight and survival rates were observed. Ruffled fur, hunched posture and lethargy were observed in TNF-α KO mice. The TNF-α KO mice had a significantly lower survival rate and greater body weight loss than WT mice (Figure 1). Within 48 h after infection, 40% of TNF-α KO mice had died. All remaining TNF-α KO mice were dead within 72 h post-infection. Within 72 h post-infection, 20% of WT mice were dead, with no further mice dying until 10 days post-infection. Losses in body weight were observed for WT and TNF-α KO mice up to 24 h post-infection. After this time point, the body weight of WT mice, while TNF-α KO mice continued to lose body weight. At 48 h post-infection, the loss in body weight for TNF-α KO mice was significant and severe.
Bacterial clearance in the lungs and blood
To examine whether TNF-α deficiency affected bacterial clearance, bacterial numbers in the lungs and blood were quantified. Bacterial counts in the lungs and blood of TNF-α KO mice were significantly higher than those in WT mice (Figure 2). Bacterial counts in the lungs of WT mice increased until 12 h post-infection, after which bacterial levels remained steady until 72 h post-infection. The bacterial counts in the lungs of TNF-α KO mice increased up to 48 h post-infection and were approximately 100-fold higher than those in WT mice. There was a large difference between bacterial counts in the blood of WT and TNF-α KO mice, with those in KO mice significantly higher. These results suggest that TNF-α might play an important role in clearing S. pneumoniae D39 and protecting the host from systemic infection.
Differential cell counts in BAL fluid
To examine white blood cell (WBC) recruitment in the BAL fluid during infection, BAL fluid was collected, and the total number of WBCs and neutrophils were quantified (data not shown). The BAL fluid from WT and TNF-α KO contained increased WBC and neutrophil counts following infection. The TNF-α KO mice contained fewer WBCs and neutrophils than those in WT mice but these differences were not significant. The proportion of neutrophils among WBCs increased in WT and TNF-α KO mice up until 48 h post-infection; however, this difference was not significant. The significant increase in bacterial numbers in the lungs of TNF-α KO mice was an unexpected result.
Histopathological lesions in the lungs and spleen
We examined lung and spleen tissue sections that had been stained with H&E for histopathological analysis. In lung sections, intra-alveolar inflammation, bronchitis, edema, and endothelialitis were observed, with the degree of pneumonia scored (Figure 3). No significant differences were observed between WT and TNF-α KO mice. These results reflect those in BAL fluid with respect to differential cell counts. In the spleen sections, those from TNF-α KO mice showed severe white pulp depletion at 48 h post-infection while spleens from WT mice had a normal appearance until 72 h post-infection (Figure 4). These results suggest that intranasal S. pneumoniae D39 infection results in systemic damage as opposed to local lung damage. Because severe white pulp depletion was observed in the spleens of TNF-α KO mice, the degree of apoptosis in white pulp was evaluated. Apoptotic cells were stained using TUNEL assays and the number of apoptotic cells counted. The white pulp of TNF-α KO mice contained a significantly increased number of apoptotic cells at 48 h post-infection. The WT mice did not exhibit such an increase until 72 h post-infection (Figure 5).
Cytokine levels in serum
To evaluate whether TNF-α deficiency affects the systemic immune response after intranasal S. pneumoniae D39 infection, IFN-γ, IL-12p70 and IL-10 levels in the serum of WT and TNF-α KO mice were measured using ELISAs. Compared with WT mice, TNF-α KO mice exhibited significantly increased IFN-γ, IL-12p70 and IL-10 levels at 48 h post-infection (Figure 6).
Discussion
The pro-inflammatory cytokine TNF-α plays a pivotal role in the regulation of immune response and inflammation. There is much evidence to indicate that TNF-α plays a crucial role in protecting the host against infectious pathogens in murine models [89] and human clinical trials [101112].
To understand the role of TNF-α in the S. pneumoniae-infected mouse model, WT and TNF-α KO mice were challenged with S. pneumoniae D39 serotype 2 and the immune response examined. The TNF-α KO mice exhibited severe clinical signs of systemic infection, along with a low survival rate. Deficiency in TNF-α induced increased bacterial numbers in the blood and lungs, especially in the blood of TNF-α KO mice. The TNF-α KO mice had bacterial counts in blood approximately 1000-fold greater than those in WT mice. These results suggest that TNF-α plays an critical role in protecting the onset of bacteremia.
Pneumolysin, an important virulence factor of S. pneumoniae, separates tight junctions between epithelial cells and promotes the production of TNF-α and IL-1 by phagocytes [1314]. It also plays an important role in the induction of sepsis [15]. The clearance of bacteria was impaired by TNF-α deficiency, while bacterial loads were increased. This was followed by increased levels of pneumolysin, possibly resulting in greater separation of tight junctions, and the induction of severe bacteremia. Histopathologically, the pneumonia scores were similar between WT and TNF-α KO mice. The neutrophil and WBC counts in BAL fluid were also similar between WT and TNF-α KO mice. Considering the early death and low survival rate of TNF-α KO mice, pneumonia was likely not the direct cause of death. Previous studies using TNF-α receptor-deficient mice and anti-TNF-α antibody-treated mice revealed minimal effects on the degree of pneumonia [1617].
Neutrophil infiltration of the lungs is a major histopathological feature of pneumococcal pneumonia, requiring TNF-α and IL-1 signaling [18]. TNF-α receptor deficiency alone is not sufficient to significantly decrease neutrophil recruitment, however TNF-α and IL-1 receptor deficiency significantly decreased neutrophil recruitment to the lungs of mice infected with S. pneumoniae serotype 3 [16].
In this study, severe sepsis and spleen damage was responsible for early death and low survival rates of TNF-α KO mice. The spleens of TNF-α KO mice exhibited severe white pulp depletion and a significantly increased number of apoptotic cells at 48 h post-infection, whereas the spleens of WT mice had a normal appearance. A similar phenomenon was also observed in a previous study. Another lethal strain of S. pneumoniae serotype 3 resulted in extensive histopathological lesions in the livers of TNF-α KO mice, however lung inflammation was lacking [17]. In a cecal puncture-induced sepsis mouse model, septicemic mice showed significantly increased levels of apoptosis and marked depletion of splenic dendritic cells. The survival ability of the host was impaired as a result of this [19]. Our results suggest that TNF-α plays a crucial immunological role in improving host resistance against sepsis and systemic organ damage that is caused by a disseminated infection.
To determine whether TNF-α deficiency affected systemic cytokine production following S. pneumoniae D39 infection, serum levels of IFN-γ, IL-12p70 and IL-10 were measured using ELISAs. IFN-γ is mainly produced by natural killer (NK) and NK T (NKT) cells, and has an important role in the immune response [20]. IL-12 is produced by dendritic cells and macrophages, and stimulates the production of IFN-γ and TNF-α [21]. IL-10 is an anti-inflammatory cytokine that limits T cell activation and suppressed pro-inflammatory responses [22]. TNF-α KO mice showed significantly increased IFN-γ, IL-12p70 and IL-10 levels at 48 h post-infection. In a previous study, IFN-γ levels in serum were significantly increased following infection with a virulent S. pneumoniae. Infection with avirulent S. pneumoniae induced much lower levels of IFN-γ in serum [23]. In the current study, a lethal D39 strain was used to infect mice; TNF-α KO mice had bacterial counts approximately 1000-fold higher in blood samples than those in WT mice. A bacterial burden that severe might induce a significant increase in IFN-γ levels in the serum of TNF-α KO mice. IL-12 is known to stimulate IFN-γ production in murine macrophages [24]. In the current study, IL-12p70 and IFN-γ levels were increased in the serum of TNF-α KO mice at 48 h post-infection. Increased IFN-γ and IL-12 production after infection is regulated by increased IL-10 levels, which acts as a regulator limiting pro-inflammatory cytokines [2526]. Our results indicate that bacterial clearance occurred, and production of inflammatory cytokines was increased to a small extent in WT mice. In contrast, the TNF-α KO mice failed to regulate an appropriate immune response against S. pneumoniae infection, resulting in increased bacterial loads and a significant increase in the levels of inflammatory cytokines.
In conclusion, TNF-α KO mice experienced continual and greater loss in body weight compared with WT mice, early death, low survival rates, clinical signs of systemic infection that correlated with impaired bacterial clearance, severe splenic damage, and increased levels of inflammatory cytokines. However, a deficiency in TNF-α levels did not significantly affect lung inflammation, and neutrophil and WBC counts. Early death and low survival rates of TNF-α KO mice are likely caused by the extent of spleen damage induced by severe bacteremia. Our findings suggest that TNF-α plays a critical role in protecting the host from a systemic S. pneumoniae infection.



 XML Download
XML Download