

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Estradiol plays very important roles in the central nervous system. For example, estradiol can modulate various activities including neuronal differentiation, synaptic transmission, anti-inflammatory, and protective actions in the development of neurodegenerative diseases [1,2,3]. Moreover, estradiol protects neurons from N-methyl-D-aspartate (NMDA)-induced neurotoxicity [4]. Estradiol also attenuates apoptotic cell death, downregulates the Bax to Bcl-2 ratio and inhibits caspase-3 activation in cortical neurons exposed to glutamate [5].
Phosphoprotein enriched in astrocytes 15 (PEA-15) is a small phosphoprotein that predominantly expressed in the central nervous system. PEA-15 was originally identified as an abundant phosphoprotein in astrocytes [6]. Moreover, previous study demonstrated the cellular localization of PEA-15 in astrocytes and neurons of brain [7]. Numerous PEA-15-immunoreactive were detected in astrocytes and neurons of the central nervous system regions [7]. PEA-15 modulates various fundamental cellular functions including cell proliferation, apoptosis, and signal integration [8,9]. PEA-15 has the N-terminal death effector domain (DED) that interacts with death receptor-activated apoptosis proteins including tumor necrosis factor (TNF) and Fas ligands [8,9,10]. PEA-15 exerts an anti-apoptotic function through the inhibition of death-inducing signaling complex formation and inactivation of the caspase cascade [10,11,12]. Moreover, PEA-15 has two phosphorylated serine residues, Ser 104 and Ser 116 [13,14,15]. Protein kinase B/Akt phosphorylates PEA-15 at Ser 116, and protein kinase C phosphorylates it at Ser 104 [13]. PKC and Akt can modulate the anti-apoptotic function of PEA-15 through phosphorylation [15]. Thus, the phosphorylation of PEA-15 is an important determinant of cell fate, apoptosis and cell survival [8,9]. We previously demonstrated the neuroprotective effects of estradiol in focal cerebral ischemia [16]. Estradiol attenuates neuronal cell death by regulation of the Akt signaling pathway [16,17]. Although many studies have shown the neuroprotective effects of estradiol, little information is available regarding the expression of the two phosphorylated forms of PEA-15 (Ser 104 and Ser 116) in the presence of estradiol during ischemic brain injury. We hypothesize that estradiol regulates the expression of PEA-15 and the phosphorylated forms of PEA-15 in ischemic brain injury. Thus, we investigated the change of these proteins by estradiol during focal cerebral ischemia and glutamate-induced neuronal cell death.
Female Sprague-Dawley rats (210-230 g, n=24) were randomly divided into four groups as follows: vehicle+sham group, estradiol+sham group, vehicle+middle cerebral artery occlusion (MCAO) group, and estradiol+MCAO group (n=6 per group). Animals were housed in standardized temperatures (25℃) and lighting (12/12 light/dark cycle). All experimental procedures for animal use were approved by the Institutional Animal Care and Use Committee of Gyeongsang National University (GNU-LA-013). Animals were bilaterally ovariectomized to remove endogenous estradiol and implanted with a silastic capsule containing vehicle or 17β-estradiol (180 g/mL, Sigma, St. Louis, MO, USA) [18]. Sesame oil (Sigma) was used as vehicle. Animals underwent surgical MCAO to induce cerebral ischemia at two weeks after ovariectomy.
MCAO was performed as previously described with some modifications [19]. Animals were injected intraperitoneally with sodium pentobarbital (30 mg/kg) for anesthetization. The right common carotid artery was exposed and all branches of the external carotid artery were isolated. The common carotid artery was clamped with vascular clips, and the external carotid artery was tied and cut. A 4/0 nylon filament heated to create a rounded tip was introduced into the external carotid artery and carefully advanced into the internal carotid artery. The filament was advanced to the base of the right middle cerebral artery. Animals were kept on a heating pad for maintenance of body temperature. Animals were decapitated and brains were rapidly removed 24 h after the onset of MCAO. Sham-operated animals underwent the same surgical procedure except the nylon filament insertion step.
The preparation of protein was performed as previously described [16,17]. The right cerebral cortex was isolated and homogenized in buffer [1% Triton X-100, 1 mM EDTA in 1×PBS (pH 7.4)] containing 200 µM phenylmethylsulfonyl fluoride and 10 µM leupeptin. The homogenates were centrifuged at 15,000 g for 20 min at 4℃ and the supernatants were collected. The protein concentration was determined using a bicinchoninic acid (BCA) kit (Pierce, Rockford, IL, USA). Total protein (30 µg) was loaded onto 10% SDS-polyacrylamide gel. The gels were electrophoresed and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% skim milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 h and washed in TBST. The membranes were incubated with the corresponding primary antibodies for 12 h at 4℃. The following antibodies were used: anti-PEA-15, anti-phospho-PEA-15 (Ser104), anti-phospho-PEA-15 (Ser116) (diluted 1:1000, Cell Signaling Technology, Beverly, MA, USA), and anti-actin (diluted 1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA). The horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (1:5000, Pierce) was used as a secondary antibody. The enhanced chemiluminescence (ECL) kit (GE Healthcare, Little Chalfont, Buckinghamshire, UK) was used to detect the signals according to the manufacturer's protocol.
Mouse hippocampal neuronal cells (HT22) were cultured in Dulbecco's modified Eagle's medium (DMEM, without L-glutamine), supplemented with 10% fetal bovine serum, streptomycin (100 µg/mL), and penicillin (100 unit/mL) (Gibco BRL, Gaithersburg, MD, USA) at 37℃ in a humidified chamber with 5% CO2 atmosphere [17]. HT22 cells were seeded on 60-mm culture dishes at 100,000 cells per dish and incubated for 24 h. We measured the cell viability depending on glutamate concentrations (1, 3, 5, 7, 10 mM) in cultured HT22 cells for the determination of glutamate concentration. Glutamate (1 or 3 mM) did not appear significantly change on cell viability in HT22 cells. However, the cell viability was decreased in neuronal cells treated with 5 mM glutamate (Supplement 1). The effect of glutamate was assessed in a dose-dependent manner in HT22 cells. Thus, we decided 5 mM concentration of glutamate for the present study. To induce oxidative stress, 5mM glutamate (Sigma) was added into the culture medium for 24 h. 17β-estradiol was treated 15 min before glutamate addition. 17β-estradiol was dissolved in 95% ethanol at a concentration of 1 mM and diluted to the final concentration (1 or 10 µM) in culture medium. It is accepted that a dose of estrogen (1 µM) protected HT22 cells from glutamate toxicity [20]. Ethanol was used as a vehicle at a final concentration of 0.1%, which has no effect on glutamate toxicity or cell viability. Cell viability was determined by measuring the metabolism of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT). Briefly, MTT solution (5 mg/mL) in serum-free medium was added to each well and applied to cells for 2 h at 37℃. Medium was removed and dimethyl sulfoxide (DMSO) was added. After shaking for 1 hat 37℃, absorbance was measured at a wavelength of 570 nm using a microplate reader. Cell survival was expressed as the percentage of neuroprotection vs. vehicle (set to100%).
We previously demonstrated that estradiol significantly reduces cerebral infarct volume and apoptotic cell death in a MCAO animal model [16]. In this study, Western blot analysis demonstrated that PEA-15 levels were decreased in vehicle+MCAO animals compared to sham-operated animals, whereas estradiol treatment prevented MCAO-induced decreases in PEA-15 expression (Figure 1A-1D). PEA-15 levels were 0.70±0.02 and 0.98±0.03 in the cerebral cortices of vehicle+MCAO and estradiol+MCAO, respectively (Figure 1B). Moreover, phospho-PEA-15 (Ser 104) and phospho-PEA-15 (Ser 116) levels were decreased in vehicle+MCAO animals. Estradiol treatment also attenuated MCAO-induced decreases in the expression of these phosphoproteins. Phospho-PEA-15 (Ser 104) levels were 0.48±0.03 and 0.94±0.03 in the cerebral cortices of vehicle+MCAO and estradiol+MCAO, respectively (Figure 1C). Phospho-PEA-15 (Ser 116) levels were 0.39±0.02 and 0.91±0.03 in the cerebral cortices of vehicle+MCAO and estradiol+MCAO, respectively (Figure 1D).
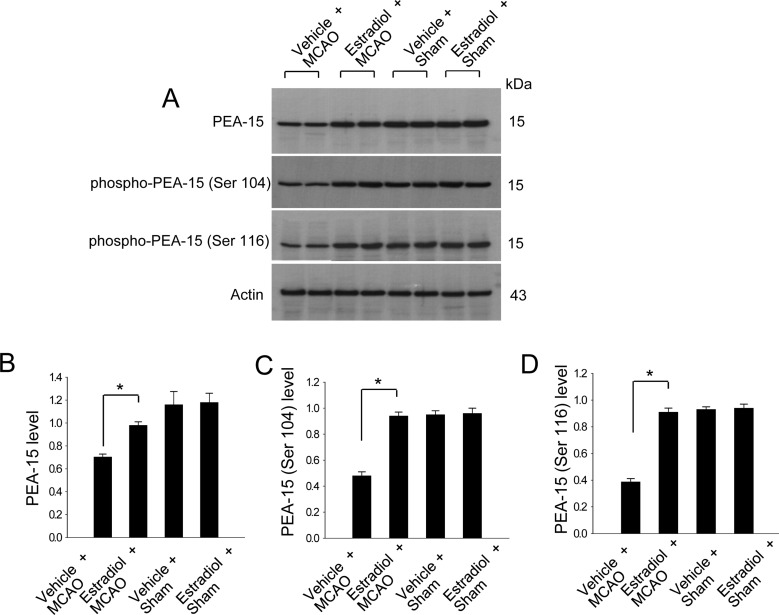
Figure 1
Western blot analysis (A-D) of PEA-15, phospho-PEA-15 (Ser 104), and phospho-PEA-15 (Ser 116) in the cerebral cortex from vehicle+MCAO, estradiol+MCAO, vehicle+sham, estradiol+sham animals. Each lane represents an individual experimental animal. Densitometric analysis is represented as intensity of these proteins to intensity of actin (B-D). Molecular weight markers (kDa) are depicted at left. Data (n=6) are represented as mean±S.E.M. *P<0.05.
![]()
In cultured neuronal cells, estradiol protected neurons against glutamate-induced toxicity (Figure 2A). Glutamate exposure decreased PEA-15, phospho-PEA-15 (Ser 104), and phospho-PEA-15 (Ser 116) levels, whereas estradiol treatment attenuated these decreases (Figure 2B). PEA-15 levels were 0.41±0.02 in the glutamatetreated group, 0.80±0.03 and 0.84±0.03 in the estradiol-treated groups (1 and 10 µM of estradiol, respectively) (Figure 2C). Phospho-PEA-15 (Ser 104) levels were 0.43±0.03 in the glutamate-treated group, 0.84±0.03 and 0.86±0.02 in the estradiol-treated groups (1 and 10 µM of estradiol, respectively) (Figure 2D). Phospho-PEA-15 (Ser 116) levels were 0.39±0.01 in the glutamate-treated group, 0.23±0.03 and 0.51±0.03 in the estradiol-treated groups (1 and 10 µM of estradiol, respectively) (Figure 2E).
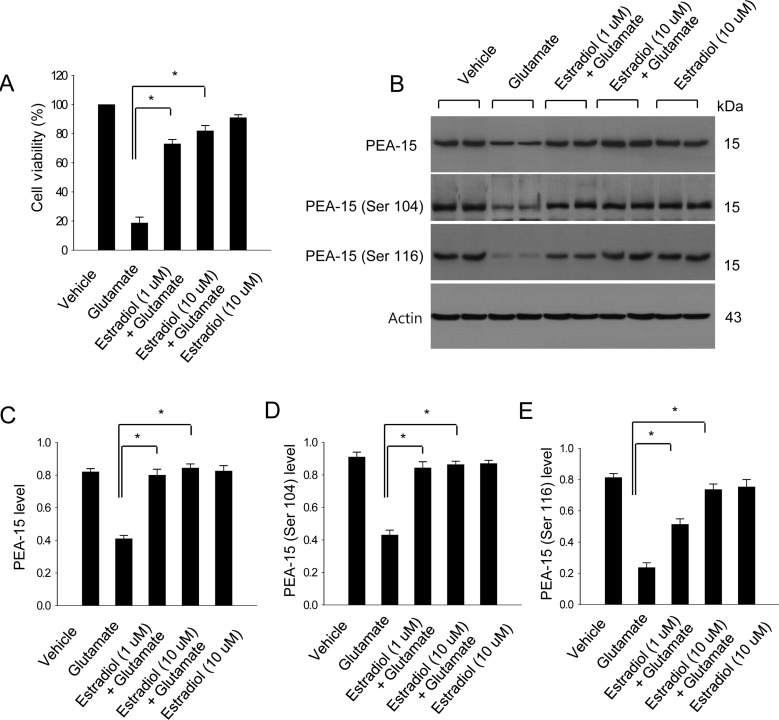
Figure 2
Cell viability (A) and Western blot analysis (B-E) of PEA-15, phospho-PEA-15 (Ser 104), and phospho-PEA-15 (Ser 116) in HT22 cells. Glutamate (5 mM) was exposed to HT22 cells for 24 h and 17β-estradiol (1 and 10 µM) treated at 15 min before glutamate exposure. Cell viability was assessed with the MTT assay (A). Cell survival was expressed as percentage of neuroprotection vs. vehicle set at 100%. Each lane represents an individual experimental animal. Densitometric analysis is represented as intensity of these proteins to intensity of actin (C-E). Molecular weight markers (kDa) are depicted at right. Data (n=5) are represented as mean±S.E.M. *P<0.05.
![]()
Estradiol has been demonstrated to preserve neuronal cells against oxidative stress [1,2]. Estradiol also prevents apoptosis against excitotoxicity in cultured neurons and exerts a neuroprotective effect [4]. Moreover, we showed that estradiol prevents neuronal cell death against ischemic brain injury by regulation of various proteins [21]. We identified a decrease in PEA-15 expression in a MCAO animal model; however, estradiol prevented this ischemic injury-induced decrease of PEA-15 expression [21]. In this study, we also showed that estradiol attenuates the ischemic injury-induced decrease in phospho-PEA-15 (Ser 104) and phospho-PEA-15 (Ser 116) protein expression.
The multifunctional protein PEA-15 is involved in the control of apoptosis and the cell cycle [9]. PEA-15 exerts a neuroprotective effect through inhibition of TNF-alpha-induced apoptosis and inactivation of the caspase cascade [9,22]. Since cerebral ischemia induces an increase of TNF-alpha and results in neuronal cell death, the inhibition of TNF-alpha reduces focal cerebral ischemic injury [23]. Moreover, ischemic injury leads to Fas-mediated neuronal cell death [24]. PEA-15 regulates apoptotic cell death by binding the Fas-associated death domain [25]. This study demonstrated that estradiol prevents ischemic injury-induced reductions in PEA-15. The preservation of PEA-15 by estradiol in injury suggests that estradiol modulates PEA-15 expression and attenuates neuronal cell death through the regulation of PEA-15 in MCAO injury.
Estradiol attenuated decreases in phosphorylated forms of PEA-15 caused by MCAO injury. PEA-15 phosphorylation is important for the modulation of cell fate including differentiation between cell death and cell survival. The phosphorylation of PEA-15 by Akt stabilizes its anti-apoptotic actions [15]. Moreover, we previously demonstrated that estradiol attenuates ischemic injury-induced reductions in Akt phosphorylation [16,17]. Our results showed that expression of phospho-PEA-15 (Ser 104) and phospho-PEA-15 (Ser 116) is reduced in MCAO injury; estradiol attenuates the reduction in these phosphorylated forms of PEA-15. As a decrease in phosphorylated forms of PEA-15 leads to apoptotic cell death, the maintenance of phospho-PEA-15 in the presence of estradiol is important for the preservation of neuronal cells in focal cerebral ischemic injury.
Glutamate toxicity leads to neuronal cell death via the induction of oxidative stress. It is accepted that glutamate induces neurotoxic effects at high concentrations [26]. In many studies, glutamate (5 mM) was used in HT22 cells for the study of neuroprotective effect against glutamate-induced apoptosis [27,28]. We measured the cell viability on various concentrations of glutamate in cultured HT22 cells. The effect of glutamate was assessed in a dose-dependent manner in cultured HT22 cells. The cell viability was significantly decreased in neuronal cells treated with 5mM glutamate. Previous study demonstrated that swollen cells were essentially not detected in cells with 2 mM glutamate, shrunken cells and swollen cells were detected in cells with higher concentrations of glutamate. We confirmed the neuroprotective effect of estradiol against glutamate-toxicity in HT22 cells. Moreover, glutamate exposure decreases the expression of PEA-15, phospho-PEA-15 (Ser 104), and phospho-PEA-15 (Ser 116) protein in HT22 cells. However, estradiol treatment prevents the glutamate toxicity-induced decrease in these proteins. We previously demonstrated that estradiol protects neuronal cells from glutamate-induced apoptosis through phosphorylation of Akt and its downstream targets, such as Bad and forkhead transcription factor [17]. This study clearly showed that estradiol regulates PEA-15 and phospho-PEA-15 (Ser 104 and Ser 116) protein expression in neuronal cells after exposure to glutamate. Taking into account the results of these in vivo and in vitro studies, estradiol has a neuroprotective function and ameliorates the ischemic injury-induced decrease of PEA-15 and phospho-PEA-15 proteins. These findings suggest that estradiol regulates phospho-PEA-15 proteins in focal cerebral ischemia and glutamate-induced neuronal cell injury, thereby attenuating cell death due to ischemic injury. In conclusion, these findings can suggest that estradiol modulates PEA-15 and phospho-PEA-15 (Ser 104 and 116) protein expression and that the regulation of these proteins contributes to the the neuroprotective effect of estradiol.
 XML Download
XML Download