

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Enolase is a multifunctional glycolytic enzyme that is expressed in various tissues and is comprised of three subunits, α, β, and γ [1]. While α-enolase is expressed in most cell types, β-enolase is expressed exclusively in muscle cells [1]. γ-Enolase is expressed only in neurons and neuroendocrine cells and is considered to be a neuron-specific enolase [1,2]. Furthermore, γ-enolase increases neuron survival and enhances neurotrophic activity [3]. γ-Enolase also promotes cell survival and neurite outgrowth by activating the phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) signaling pathways [4].
Ferulic acid (4-hydroxy-3-methoxycinnamic acid) is found in Anglica sinensis (Olivi.) Didl and Ligusticum chuoanxiong Hort; these plants have traditionally been used as Chinese herbal medicines for treating stroke. Ferulic acid has been shown to reduce cerebral ischemic injury and to provide neuroprotection against middle cerebral artery occlusion (MCAO) [5,6,7]. Ferulic acid has also been shown to inhibit glutamate-induced apoptosis in cultured cortical neurons by regulating the PI3K and extracellular signal-regulated kinase (ERK) signaling pathways [8]. Moreover, ferulic acid has been shown to exert various therapeutic effects, including anti-inflammatory, anti-oxidant, anti-aging, and neuroprotective effects [5,9,10,11,12]. Although ferulic acid exerts a neuroprotective effect in ischemic injury, the mechanism by which ferulic acid exerts this effect remains unclear. Using a proteomics approach, we found that focal cerebral ischemia decreases γ-enolase expression, whereas ferulic acid attenuates this injury-induced decrease. The aim of this study was to investigate whether ferulic acid regulates γ-enolase expression in focal cerebral ischemic injury and glutamate exposure-induced neuronal cell death.
Materials and Methods
Adult male Sprague-Dawley rats (210-230 g, n=52) were housed under controlled temperature (25℃) and lighting (12/12 h light/dark cycle) conditions and given free access to food and water. The Institutional Animal Care and Use Committee of Gyeongsang National University approved all experimental protocols for animal use (GNU-120806-R0034). Animals were randomly divided into four groups as follows: vehicle + sham group, ferulic acid + sham group, vehicle + MCAO group, and ferulic acid + MCAO group (n=13 per group). Animals were used for proteomics study (n=3 per group), used for RT-PCR analysis analysis (n=5 per group), and used for Western blot analysis (n=5 per group). Ferulic acid (Sigma, St. Louis, MO, USA) was dissolved in saline solution and injected intravenously (100 mg/kg) immediately after MCAO [5]. Saline solution was used as the vehicle.
Right MCAO was induced using a standard surgical operation as previously described [13]. Briefly, rats were anesthetized with sodium pentobarbital (30 mg/kg, intraperitoneal injection) before MCAO. Then, the right common carotid artery, external carotid artery, and internal carotid artery were exposed through a midline incision. A 4/0 monofilament nylon suture with rounded tip by heating was inserted into the right external carotid artery and gently advanced into the internal carotid artery until it blocked the origin of the middle cerebral artery. Body temperature was maintained at approximately 37℃ with a heating pad throughout the surgical procedure. Sham-operated animals underwent the same surgical procedure, except that they did not receive a monofilament nylon insertion. At 24 h after occlusion, animals were sacrificed and right cortex tissues were isolated.
A proteomics approach was carried out as previously described [14]. Briefly, samples were initially dissolved in lysis buffer (8 M urea, 4% CHAPS, ampholytes, and 40 mM Tris-HCl). After centrifugation of the lysates at 16,000 g for 20 min at 4℃, the supernatants were collected, and the total protein concentrations were determined with the Bradford assay (Bio-Rad, Hercules, CA, USA). Then, 50 µg protein was mixed with rehydration solution, and isoelectric focusing (IEF) was performed for 13 h using immobilized pH gradient gel strips (IPG, pH 4-7, pH 6-9, 17 cm, Bio-Rad). Proteins were focused on Protean IEF Cells (Bio-Rad) at 20℃ using the following conditions: 250 V (15 min), 10,000 V (3 h), and then a ramp from 10,000 to 50,000 V. After IEF, IPG strips were applied to gradient gels (7.5-17.5%) and electrophoresed at 10 mA/gel for 10 h using Protein-XI electrophoresis equipment (Bio-Rad) at 10℃. Gels were then fixed in a solution containing 12% acetic acid and 50% methanol for 2 h and subsequently stained with silver stain solution (0.2% silver nitrate and 0.75 mL/L formaldehyde). Silver-stained gels were then scanned using an Agfar ARCUS 1200™ scanner (Agfar-Gevaert, Mortsel, BEL) and were analyzed with PDQuest software (Bio-Rad) according to a standard protocol. Protein spots were subjected to trypsin digestion and the resultant peptides were extracted using extraction buffer (5% trifluoroacetic acid in 50% acetonitrile). Extracted peptides were dried using a vacuum centrifuge for 20 min and the resultant samples were analyzed with a Voyager-DE™ STR biospectrometry workstation (Applied Biosystems, Forster City, CA, USA) for MALDI-TOF mass spectrometric analysis. Database searches were carried out using MS-Fit and ProFound programs. SWISS-PROT and NCBI were used as the protein sequence databases.
Total RNA was extracted using Trizol reagent according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). Total RNA (1 µg) was reverse transcribed for RT-PCR using the Superscript First-Strand System (Invitrogen) according to the manufacturer's protocol. Primer sequences used to amplify γ-enolase were 5'-TGGATCTCCATACTGCCAAAG-3' (forward) and 5'-CCAACTCCTCTTCAATCCTCAT-3' (reverse). Primer sequences used to amplify β-actin were 5'-GGGTCAGAAGGACTCCTACG-3' (forward) and 5'-GGTCTCAAACATGATCTGGG-3' (reverse). Thirty cycles of amplification were performed under the following conditions: melting at 95℃ for 30 sec, annealing at 52.5℃ for 30 sec, and extension at 72℃ for 30 sec. PCR products were resolved on 1% agarose gels and visualized under UV light.
For Western blot analysis, right cerebral cortices were quickly removed and homogenized in lysis buffer [1% Triton X-100 and 1 mM EDTA in PBS (pH 7.4)] containing 10 mM leupeptin and 200 µM phenylmethylsulfonyl fluoride. Lysates were clarified by centrifugation at 16,000 g for 20 min at 4℃, and the resultant supernatants were collected. Protein concentrations of the supernatants were determined using the bicinchoninic acid assay (Pierce, Rockford, IL, USA). Proteins (30 µg) were resolved on 10% SDS-polyacrylamide gels and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). Membranes were incubated with 5% skim milk and incubated with the following antibodies: rabbit polyclonal anti-rat γ-enolase (diluted 1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and mouse monoclonal anti-rat actin (diluted 1:1000, Santa Cruz Biotechnology). Membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (diluted 1:5000, Pierce, Rockford, IL, USA). An enhanced chemiluminescence Western blot detection kit (Amersham Pharmacia Biotech, Piscataway, NJ, USA) was used for detection of immunoreactive bands according to the manufacturer's protocol. Band intensities were analyzed with SigmaGel 1.0 (Jandel Scientific, San Rafael, CA, USA) and SigmaPlot 4.0 (SPSS Inc., Point Richmond, CA, USA).
The mouse hippocampal cell line HT22 was cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco BRL, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum and streptomycin/penicillin (100 µg/mL and 100 units/mL, respectively). Cells were grown at 37℃ in a humidified incubator with a 5% CO2 atmosphere. HT22 cells were seeded on 60-mm culture dishes at 100,000 cells per dish and incubated for 24 h prior to treatment. Glutamate (Sigma) was diluted to a final concentration of 5 mM in the culture medium and cells were exposed for 24 h. Ferulic acid (1 or 2.5 mM) was added to cells 1 h before glutamate addition. Cell viability after treatment was assessed with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay. Briefly, MTT solution (5 mg/mL) was added to the culture medium and cells were maintained for 4 h at 37℃. After this incubation, the MTT solution was removed and a solubilization solution containing 20% sodium dodecyl sulfate (pH 4.8) and 50% dimethylformamide was added. After an overnight incubation, absorption values at 570 nm were measured. Cell viability was then expressed as a percentage of control cell viability (set to 100%).
All data are expressed as mean±SEM. Results from each group were compared by one-way analysis of variance (ANOVA) followed by a post-hoc Scheffe's test. Differences were considered significant when P<0.05.
Go to : 
Results
We previously demonstrated that treatment with ferulic acid decreases infarct volume and apoptotic cell death after MCAO-induced brain injury [6,7,15]. Figure 1 shows that γ-enolase level decreased in the cerebral cortices of animals in the vehicle + MCAO group. However, this decrease in γ-enolase expression was attenuated in animals in the ferulic acid + MCAO group, with levels of γ-enolase remaining similar to those of sham-operated animals. γ-Enolase levels were 1.00±0.01 and 1.01±0.02 in the vehicle + sham and ferulic acid + sham groups, respectively. Moreover, γ-enolase levels were 0.31±0.02 and 1.03±0.03 in the vehicle + MCAO and ferulic acid +MCAO groups, respectively.
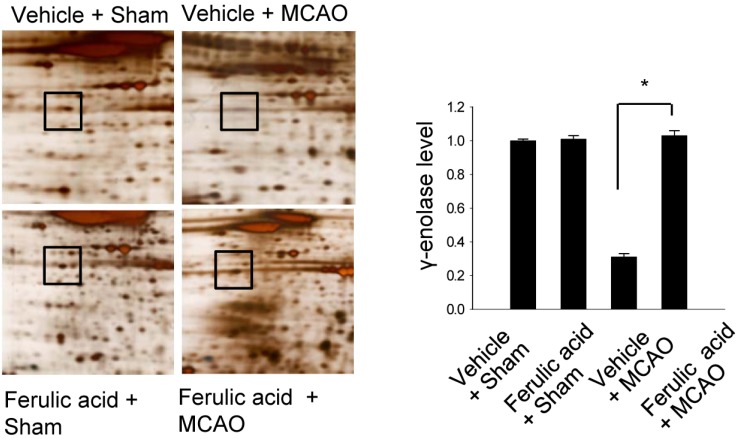 | Figure 1γ-Enolase protein spots identified by MALDI-TOF in the vehicle + sham, ferulic acid + sham, vehicle + middle cerebral artery occlusion (MCAO), ferulic acid + MCAO groups. Squares indicate the γ-enolase protein spots. Spot intensities were measured using PDQuest software. Intensity ratios represent the average spot intensities of treated animals relative to the average spot intensity of control (vehicle + sham) animals. Data (n=3) are shown as mean±SEM. *P<0.05.
|
To validate this result with a complementary technique, we performed RT-PCR analysis and confirmed that ferulic acid prevents the MCAO-induced decrease of γ-enolase expression (Figure 2). The transcript levels of γ-enolase were expressed at similar levels in the sham-operated groups. The transcript levels of γ-enolase were 0.98±0.02 and 0.99±0.03 in the vehicle + sham and ferulic acid + sham groups. The transcript levels of γ-enolase were 0.75±0.03 and 0.97±0.02 in the vehicle + MCAO and ferulic acid + MCAO groups, respectively. Moreover, Western blot analysis also showed that γ-enolase protein levels were reduced in animals in the vehicle + MCAO group compared with sham-operated animals, whereas ferulic acid treatment prevented this MCAO-induced reduction in γ-enolase protein level (Figure 3). In sham-operated groups, γ-enolase protein levels were 0.86±0.04 and 0.84±0.02 in the vehicle- and ferulic acid-treated animals. The γ-enolase protein levels were 0.67±0.02 and 0.82±0.03 in the vehicle + MCAO and ferulic acid +MCAO groups, respectively.
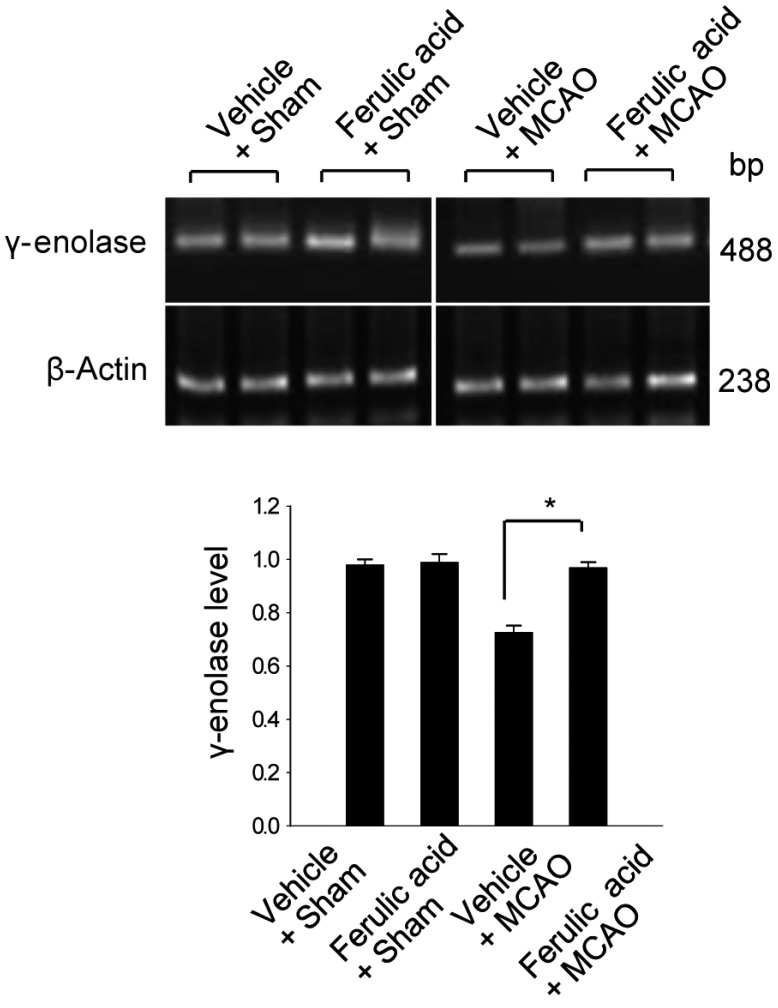 | Figure 2Reverse-transcription PCR analysis of γ-enolase expression levels in the vehicle + sham, ferulic acid + sham, vehicle + middle cerebral artery occlusion (MCAO), ferulic acid + MCAO groups. Each lane represents an individual animal. Densitometric analysis was performed to express the intensity of each γ-enolase band relative to the intensity of the corresponding actin control band. Data (n=5) are shown as mean±SEM. *P<0.05.
|
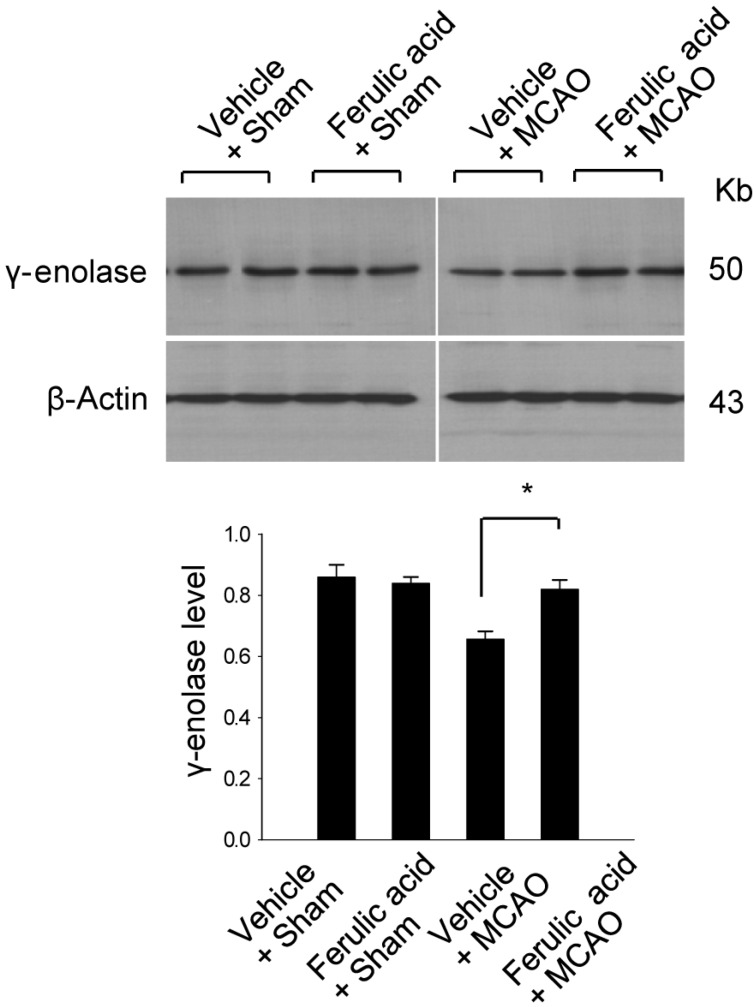 | Figure 3Western blot analysis of γ-enolase protein levels in the vehicle + sham, ferulic acid + sham, vehicle + middle cerebral artery occlusion (MCAO), ferulic acid + MCAO groups. Each lane represents an individual animal. Densitometric analysis was performed to express the intensity of each γ-enolase band relative to the intensity of the corresponding actin control band. Data (n=5) are shown as mean±SEM. *P<0.05.
|
We examined the action of ferulic acid in vitro study using glutamate-treatment to induce cell death in HT22 cells. We found that ferulic acid treatment prevented glutamate-induced cell death. In accordance with our in vivo data, γ-enolase expression was decreased in glutamatetreated HT22 cells, whereas ferulic acid treatment prevented this glutamate-induced decrease of γ-enolase expression (Figure 4). The level of γ-enolase was 0.59±0.02 in the glutamate only-treated group, and 0.86±0.02 and 0.88±0.03 in the glutamate plus ferulic acid-treated groups (1 and 2.5 mM ferulic acid, respectively).
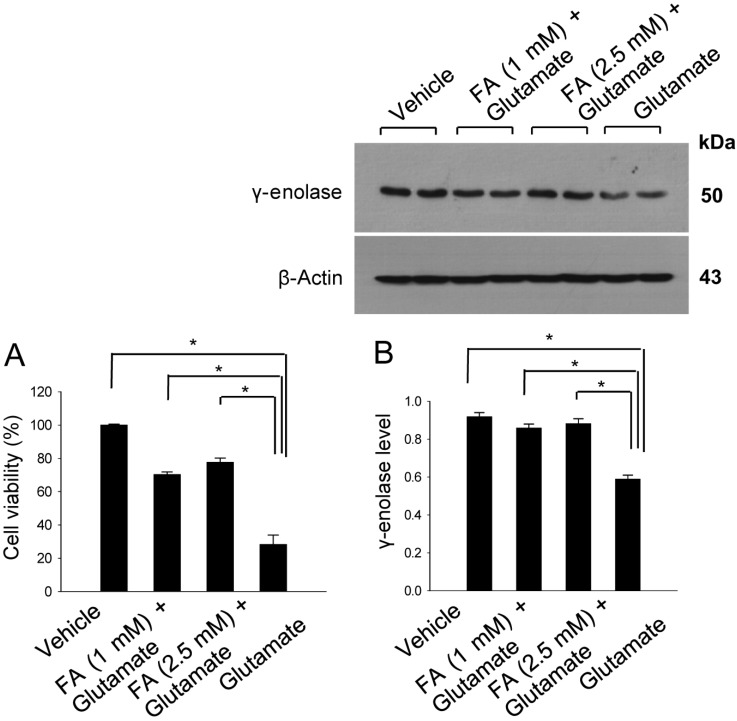 | Figure 4Cell viability (A) and Western blot analyses of γ-enolase protein levels (B) in hippocampal neuronal HT22 cells. Glutamate (5 mM) was added to HT22 cells for 24 h, with ferulic acid (FA, 1 or 2.5 mM) added to cells 30 min before glutamate exposure. Cell viability was assessed using the MTT assay (A). Cell survival is expressed as the percentage of viable treated cells out of the percentage of viable vehicle-treated cells (set to 100%). Densitometric analysis was performed to express the intensity of each γ-enolase band relative to the intensity of the corresponding actin control band (B). Data (n=5) are represented as mean±SEM. *P<0.05.
|
Go to :
Discussion
Ferulic acid exerts a strong neuroprotective effect in focal cerebral ischemia and preserves brain tissue against MCAO-induced apoptotic cell death [5]. Furthermore, ferulic acid reduces infarct volume in focal cerebral ischemia through its anti-inflammatory and anti-oxidant actions [5,9]. We previously demonstrated that ferulic acid decreases infarct volume and protects neuronal cells in an animal model of MCAO [15]. In this study, we showed that MCAO injury decreases the expression of γ-enolase in vivo, whereas ferulic acid treatment attenuates this decrease. Moreover, we also showed that glutamate treatment decreases γ-enolase expression in an in vitro model of injury, ferulic acid attenuates this decrease in γ-enolase expression.
It is well-established that γ-enolase promotes neuron survival and enhances neurite regeneration [3]. Moreover, it is known that γ-enolase regulates neuron survival and differentiation by activating the PI3K/Akt and MAPK transduction pathways [4]. Consistent with these actions, γ-enolase acts as a neurotrophic factor and exerts neuroprotective effects [2,3]. Ferulic acid has also been shown to exert a neuroprotective effect against glutamate toxicity in cortical neurons through a mechanism involving modulation of PI3K and ERK signaling pathways [8]. Moreover, our previous study demonstrated that ferulic acid prevents neuronal cell death following MCAO and these protective effects are due to inhibition of Akt signaling pathway inactivation [16]. In this study, our results clearly show that ferulic acid modulates γ-enolase expression in both MCAO-induced ischemic brain injury and glutamate-induced neuronal cell damage. Thus, our data suggest that ferulic acid maintains the level of γ-enolase expression in an animal model of MCAO and the maintenance of γ-enolase mediates the neuroprotective action of ferulic acid. However, further studies are required to more completely elucidate the mechanisms relating ferulic acid with γ-enolase expression. In conclusion, ferulic acid regulates γ-enolase expression during brain ischemia, and that modulation of γ-enolase by ferulic acid contributes to its neuroprotective effects.
Go to :
 XML Download
XML Download