

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Stroke is a major cause of death and declines the quality of life, and is characterized by neurological symptoms caused by ischemia after vascular incidents [1]. Cerebral ischemia caused by occlusions of arteries in the brain accounts for more than 80% of all stroke cases [2]. Cerebral ischemia leads to increases of intracellular Ca2+ and mitochondrial bioenergetics dysfunction, which in turn results in neuronal cell death due to the activation of enzymes that generate reactive oxygen species (ROS) [3]. The intracellular Ca2+ overload triggers cell death programs, whereas inhibitors of intracellular Ca2+ influx attenuate mitochondrial damage and preserve neuronal cells from ischemic injury [4-6]. Thus, intracellular Ca2+ overloading is an essential event in ischemic brain injury.
Glutamate is a major neurotransmitter that plays an important role for learning and memory in the central nervous system [7,8]. However, excessive production of glutamate leads to neurotoxicity and neuronal degeneration. Glutamate neurotoxicity is involved in the neurodegenerative disorders, including stroke, Alzheimer's and Parkinson's diseases. It has been reported that the levels of glutamate in neurodegenerative diseases patients are significantly higher than those in normal controls [9]. Glutamate exposure induces an increase of neuronal Ca2+ influx and leads to neuronal death. The termination of glutamate exposure decreases extracellular Ca2+ concentration and reduces neuronal degeneration [10].
Calcium binding proteins play critical roles in physiological processes, signal transduction, and muscle contraction [11,12]. Calcium binding proteins protect neurons against damage caused by excessive Ca2+ elevation. Calbindin is a calcium binding protein that contains EF-hand domains. Calbindin is considered a critical factor that performs multiple functions in neuronal cell physiology [12]. Moreover, calbindin binds calcium and modulates a second messenger molecule within cells [12]. Calbindin exists in neuron and astrocytes of the nervous system [12]. Calbindin regulates calcium homeostasis, inhibits neurodegeneration, and protects cells from apoptosis [13,14]. Cerebral ischemic injury leads to neuronal cell death through protein regulation and the modulation of complex mechanisms. However, little information is available regarding calbindin expression after brain ischemia. This study investigated calbindin expression in a middle cerebral artery occlusion (MCAO)-induced cerebral ischemia rat model and in glutamate-exposed hippocampal neurons.
Materials and Methods
Experimental animals
Male Sprague-Dawley rats (200-220 g, n=30) were randomly divided into two groups, sham-operated group and MCAO-operated group (n=10 per group). Animals were kept under temperature (25℃) and lighting (12/12 light/dark cycle). All experimental procedures for animals were approved by Institutional Animals Care and Use Committee at Gyeongsang National University (GNU-LA-015).
Middle cerebral artery occlusion
Focal cerebral ischemia was employed by middle cerebral artery occlusion (MCAO), and MCAO was performed as in a previously described method [15]. Animals were anesthetized with sodium pentobarbital (30 mg/kg, intraperitoneal injection). The right common carotid artery was exposed and the external carotid artery was cut. A 4/0 nylon suture with its tip rounded by heat was introduced into the right external carotid artery and advanced into the right internal carotid artery until the tip occluded the origin of the right MCA. Animals were scarified 24 h after the onset of MCAO.
Triphenyltetrazolium chloride (TTC) staining
Brain tissues (n=5 per group) were carefully removed from skull and cut into coronal slices of 2 mm in thickness using brain matrix. These slices (bregma -1.4~0.6 mm) were incubated for 20 min in a 2% triphenyltetrazolium chloride (TTC; Sigma, St. Louis, MO, USA) solution and fixed in 10% formalin solution. The stained tissues were scanned by an Epson Perfect V700 (Epson, Suwa, Nagano, Japan) and measured for the ischemic lesion by Image-ProPlus 4.0 software (Media Cybernetics, Silver Spring, MD, USA). The ischemic lesion percentage of each slice was calculated by the ratio of the infarction area to the whole slice area.
RNA isolation and reverse transcription-PCR amplification
The ischemic regions of right cerebral cortices (n=5 per group) were collected and were quickly frozen. Total RNA was extracted using Trizol reagent according to the manufacturer's instruction (Invitrogen, Carlsbad, CA, USA) and RNA (1 µg) was reverse transcribed using superscript first-strand system for RT-PCR (Invitrogen) based on the manufacturer's protocol. The primers sequences of calbindin are 5'-GATGCCAGCAACTGA AGT-3' (forward primer) and 5'-GGCCTAAGCATAGA CTTT-3' (reverse primer), which yielded a PCR product of 732 bp. Moreover, the primers sequences of actin are 5'-GGGTCAGAAGGACTCCTACG-3' (forward primer) and 5'-GGTCTCAAACATGATCTGGG-3' (reverse primer), which yielded a PCR product of 238 bp. The PCR reaction was carried out as followed: 5 min at 94℃, 30 sec at 94℃, 30 sec at 54℃, 1 min at 72℃ and 10 min at 72℃. The samples were amplified 30 cycles. PCR product was run in a 1% agarose gel and visualized under UV light.
Western blot analysis
Western blot analysis was performed as previously described with some modification [16,17]. The ischemic regions of right cerebral cortices (n=5 per group) were lysed in lysis buffer [1% Triton X-100, 1 mM EDTA in 1×PBS (pH 7.4)] containing 10 µM leupeptin and 200 µM phenylmethylsulfonyl fluoride. The lysates were centrifuged at 15,000 g for 20 min at 4℃ and the supernatants were collected. The protein concentration was measured using the bicinchoninic acid (BCA) kit (Pierce, Rockford, IL, USA) according to the manufacturer's protocol. Total proteins (30 µg) were loaded on 10% SDS-polyacrylamide gel electrophoresis and were transferred to a poly-vinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA, USA). Immunoreaction was carried out using the following antibodies: anti-calbindin (diluted 1:1000, Cell Signaling Technology, Beverly, MA, USA) and anti-actin (diluted 1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA). The membranes were washed in Tris-buffered saline containing 0.1% Tween-20 (TBST) and were sequentially reacted with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (1:5000, Cell Signaling Technology) for calbindin, HRP-conjugated anti-mouse IgG (1:5000, Cell Signaling Technology) for actin. The immunoreactive bands were detected by chemiluminescence using the ECL Western blot analysis system (Amersham Pharmacia Biotech, Piscataway, NJ, USA). The intensity analysis was carried out using SigmaGel 1.0 (Jandel Scientific, San Rafael, CA, USA) and SigmaPlot 4.0 (SPSS Inc., Point Richmond, CA, USA).
Immunohistochemical staining
Immunohistochemistry was performed as in a previously described method [17]. Brain tissues (n=5 per group) were fixed in 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS, pH 7.4) and embedded in paraffin. The paraffin sections were blocked with 1% normal goat serum in PBS for 1 hour, and then reacted with anti-calbindin antibody (diluted 1:100, Cell Signaling Technology) at 4℃ for 15 hours. After washing with PBS, sections were consecutively reacted with biotin-conjugated goat anti-rabbit IgG (1:200 in PBS) for 1 h, followed by incubation with an avidin-biotin-peroxidase complex for 1 h from a Vector ABC Elite kit (Vector Laboratories Inc., Burlingame, CA, USA). Brain sections were washed with PBS and stained with diaminobenzidine tetrahydrochloride (Sigma) solution with 0.03% hydrogen peroxidase for 3 min. Sections were counterstained with hematoxylin and dehydrated in graded alcohol. Slides were observed under a microscope and then photographed. The total cell number and calbindin-positive cell number were obtained in the ischemic core region. The index of positive cells was described as the percentage of the number of calbindin-positive cells to the total number of cells.
Cell culture and treatment
Mouse hippocampal cell lines (HT22) were cultured as in a previously described method [12,13,14]. HT22 cells were cultured in Dulbecco's modified Eagle's medium (DMEM, without L-glutamine) with 10% fetal bovine serum, streptomycin (100 µg/mL), and penicillin (100 unit/mL) (Gibco BRL, Gaithersburg, MD, USA). HT22 cells were incubated in a humidified incubator with 5% CO2 at 37℃, and were seeded on 60-mm culture dishes at 100,000 cells per dish. Cell density was maintained 70% or less confluency to attenuate excessive growth [18]. Glutamate (Sigma) was treated with a final concentration of 5 mM in culture medium and cells were maintained for 24 h. Cell viability was investigated by measuring metabolism of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) [19]. MTT solution (5 mg/mL) was added to the culture medium, HT22 cells were maintained for 4 h at 37℃, and then MTT solution were removed. The solubilization solution containing 20% sodium dodecyl sulfate (pH 4.8) and 50% dimethylformamide was added and absorption at 570 nm were measured. Cell viability was determined as percentage of living cells vs. vehicle set at 100%.
The measurement of intracellular Ca2+ concentration
The intracellular Ca2+ concentration was measured by calcium indicator Fura-2/AM [20]. The cells were incubated in the DMEM media containing 10 µM fura-2/AM at 37℃ for 1 h. Fura-2 fluorescence signals of cells were measured by luminescence spectrophotometer (LS50B, Perkin Elmer, Boston, MA, USA). The Ca2+ fluorescence was recorded at excitation wavelengths of 340 and 380 nm, the fluorescence emitted at 510 nm was revealed by a photon-counting photomultiplier. The spectrum of fura-2/AM released a peak at 480 nm. Fluorescence signals were analyzed by a MicroVax II computer and software (Origin 7). The intracellular Ca2+ concentration was determined using the ratio method as in a previously described equation [21].
Statistical analysis
In data of RT-PCR and Western blot analyses, calbindin levels are represented as intensity of calbindin to intensity of actin. All data are expressed as mean±SEM. The results in each group were compared by Student's t-test. The difference for comparison was considered significant at *P<0.05.
Go to : 
Results
MCAO-operated group significantly increases infarct volumes according to TTC staining (Figure 1A). The infarct regions were 2.3±0.9% and 29.3±2.8% in sham-operated and MCAO-operated groups (Figure 1B). Calbindin expression was decreased in MCAO-operated groups compared to sham-operated groups. In result of RT-PCR, calbindin levels were 0.87±0.03 and 0.58±0.04 in the cerebral cortices of sham-operated and MCAO-operated groups, respectively (Figure 1C, 1D). Moreover, calbindin protein levels were 0.94±0.05 and 0.82±0.05 in the cerebral cortices of sham-operated and MCAO-operated groups, respectively (Figure 2A, B). Immunohistochemical staining showed that the numbers of positive cells for calbindin were decreased in the cerebral cortices of vehicle+MCAO animals relative to sham-operated groups (Figure 2C, 2D). The index of positive cells were 33.8±2.2% and 10.2±2.4% in sham-operated animal and vehicle+MCAO, respectively (Figure 2E).
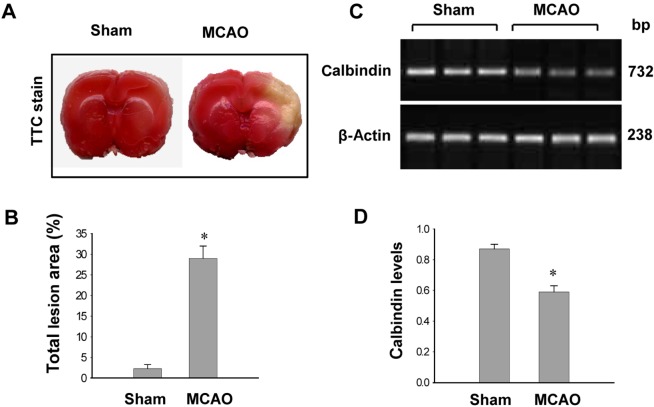 | Figure 1Photographs of TTC staining (A and B) and RT-PCR data of calbindin (C and D) in the sham-operated and middle cerebral artery occulssion (MCAO)-operated animals. The ischemic area appeared white, while the intact area appeared red (A). The percentage of ischemic lesion area was calculated by the ratio of the infarction area to the whole slice area (B). Densitometric analysis is represented as intensity of calbindin to intensity of actin (D). Data (n=5) are shown as mean±SEM. *P<0.05 (vs. Sham).
|
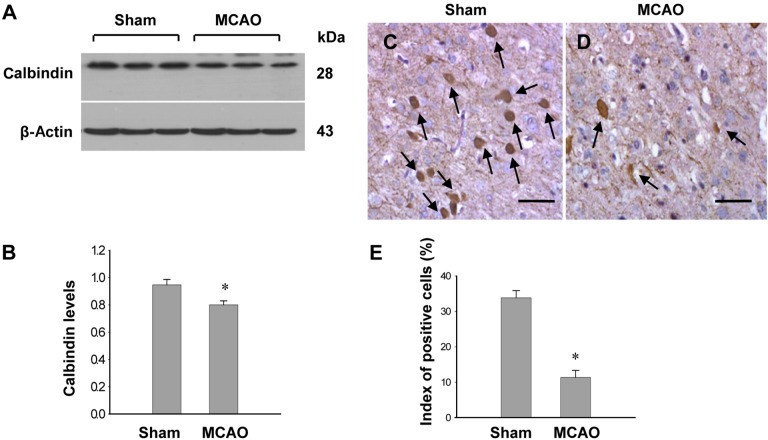 | Figure 2Western blot analysis of calbindin (A and B) and immuno-staining of calbindin (C and D) in the sham-operated and middle cerebral artery occulssion (MCAO)-operated animals. Densitometric analysis is represented as intensity of calbindin to intensity of actin. Data (n=5) are shown as mean±SEM. *P<0.05 (vs. Sham). Arrows indicate positive cells of calbindin. The index of positive cells indicated the percentage of calbindin-positive cells (E). Scale bar=100 µm.
|
Glutamate toxicity produces oxidative stress and induces neuronal cell death in HT22 cells. Glutamate exposure increases cell death in cultured HT22 cells and significantly increased intracellular Ca2+ concentrations (Figure 3A, 3B). Western blot analysis showed that glutamate exposure induces decreases of calbindin in HT22 cells (Figure 3C). Calbindin protein levels were 0.76±0.04 and 0.58±0.05 in the vehicle- and glutamate-treated group, respectively (Figure 3D).
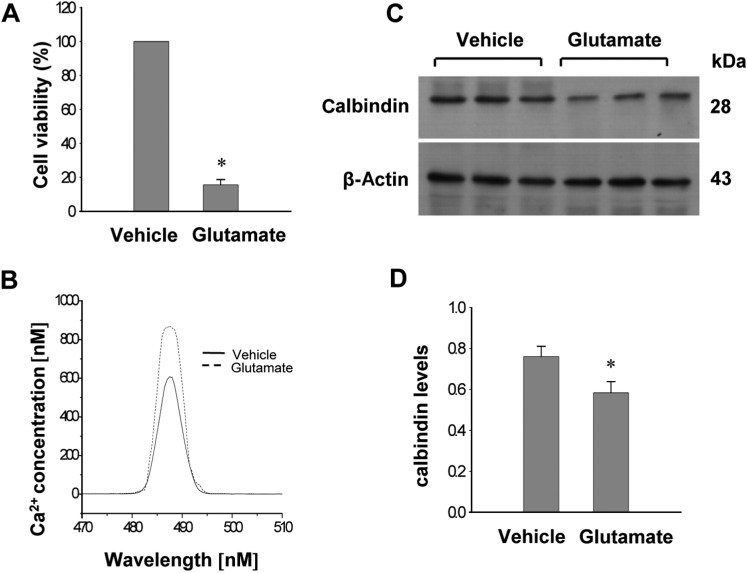 | Figure 3Cell viability (A), intracellular calcium concentration (B), Western blot analysis of calbindin (C and D) in HT22 cells. Glutamate (5 mM) or vehicle was exposed to HT22 cells for 24 h. Cellular viability was assessed using the MTT assay (A). Cell viability was expressed as percentage of neuroprotection vs. vehicle set at 100%. Neuronal cell are followed by fura-2/AM labeling, and fluorescence spectra for calcium are measured by luminescence spectrophotometer (B). Densitometric analysis is represented as intensity of calbindin to intensity of actin (D). Data (n=5) are represented as mean±SEM. *P<0.05 (vs. Vehicle).
|
Go to :
Discussion
Focal cerebral ischemia caused by MCAO leads to neuronal cell death and ischemic infarctions [15]. This study demonstrated serious infarct damages in the cerebral cortices of MCAO-operated animals and reductions of calbindin levels in MCAO-induced brain injury. Ischemia-induced hypoxic injury triggers the activation of degradative enzymes including phospholipases and proteases [5]. The activations of these enzymes change membrane permeability and induce increases of intracellular Ca2+, leading to the disruption of cell calcium homeostasis [5,6]. The calcium overload can induce caspase activation and cell death [6]. Moreover, calcium overload is observed in ischemic neuronal death [6]. Therefore, calcium overload is accepted as one of the main triggers of neuronal cell death. Over-expression of calbindin reduces oxidative stress and preserves mitochondrial function in Alzheimer's disease model [22]. Mover, calbindin over-expression protects neurons from focal cerebral ischemia and protein transduction domain (PTD)-calbindin D 28k pretreatment attenuates rat brain injury induced by ischemia and reperfusion [23,24]. RT-PCR and Western blot analyses showed decreases in calbindin protein during MCAO-induced brain injury. Immunohistochemical staining clearly showed decreases of calbindin immunoreactivity in MCAO-induced brain injury. The decreases of calbindin in cerebral ischemia cause the disruption of calcium homeostasis and leads to neuronal cell death.
Glutamate toxicity leads to serious oxidative stress and also results in neuronal cell death [18]. Glutamate exposure significantly decreases cell viability and increases apoptotic cell death in cultured hippocampal cells [16]. This study showed that glutamate treatment induces increases in intracellular Ca2+ concentrations and decreases in calbindin expression. Increases in intracellular calcium concentration lead to the disintegration of cells through the activation of the caspase cascade, consequently leading to neuronal cell death [3]. Thus, low calcium binding protein concentrations contribute to the imbalance of calcium homeostasis, and lead to neuronal dysfunction [10]. Moreover, over-expression of calbindin prevents apoptosis in cultured neuronal cells expressing mutant presenilin 1 that causally linked to early-onset inherited forms of Alzheimer's disease [22]. Low calbindin concentrations indicate the imbalance of calcium homeostasis and neuronal dysfunctiom. This study showed that calbindin levels are decreased in glutamate-exposed HT22 cells. In conclusion, in vivo and in vitro studies indicate the decrease of calbindin in ischemic brain injury, and the increase of intracellular calcium levels in glutamate-exposed neuronal cells damage. Taken together, these findings suggest that decreases of calbindin induce imbalances of calcium homeostasis, leading to neuronal cell death in MCAO-induced injury and glutamate-exposed damage.
Go to :
 XML Download
XML Download