

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Gingko biloba extract 761 (EGb 761) is extracted from the green leaves of Gingko biloba. EGb 761 mainly consists of flavonoid glycosides and unique terpene lactones that exhibit anti-oxidant properties in the ischemic brain [1,2]. EGb 761 has been utilized as a traditional therapy and has therapeutic effects in the treatment of Alzheimer disease and vascular disorders [3-5]. EGb 761 acts as a scavenger of free radicals and protects neuronal cells against ischemic injury [6].
Hippocalcin is a calcium buffering protein that belongs to the EF-hand Ca2+ binding protein family. Hippocalcin plays an important role in the maintenance of Ca2+ homeostasis in the central nervous system because it binds to Ca2+, which is released into the cytoplasm [7-10]. The excitotoxic reactions of excessive calcium in neurons lead to apoptotic cell death and result in central nervous system disorders [11]. Hippocalcin exerts a neuroprotective effect through the elimination of excessive intracellular calcium. Although many studies have suggested the neuroprotective effects of EGB 761, further studies are necessary to completely understand its neuroprotective mechanism [12,13]. We investigated whether EGb 761 regulates the expression of hippocalcin during cerebral ischemic injury.
All animal experiments were followed a protocol approved by the Committee for Animal Experimentation at the Gyeongsang National University (GNU-LA-13). Male Sprague-Dawley rats (200-220 g, n=60) were purchased from Samtako Co. (Animal Breeding Center, Osan, Korea). Animals were randomly divided into four groups, vehicle+sham group, EGb 761+sham group, vehicle+middle cerebral artery occlusion (MCAO) group, and EGb 761+MCAO group (n=15 per group). EGb 761 (100 mg/kg, Yuyu, Seoul, Korea) was dissolved in normal saline and treated via an intraperitoneal injection at 1 h before the onset of MCAO [13]. The same volume of normal saline was given as the vehicle.
Focal cerebral ischemia model was conducted by performing MCAO, as previously described method [14]. Briefly, animals were anesthetized with sodium pentobarbital (30 mg/kg) and the right common carotid artery was exposed through a midline cervical incision. The external carotid artery was tied and cut. A 4/0 nylon suture with its tip rounded by heating over a flame was inserted into the external carotid artery and advanced into the internal carotid artery, thereby occluding blood flow through the middle cerebral artery. At 24 h after the onset of permanent occlusion, brain tissues were removed. Sham-operated animals underwent the same surgical procedure without suture insertion.
The right cerebral cortices were lysed in buffer (8M urea, 4% CHAPS, ampholytes, and 40 mM Tris-HCl) and centrifuged at 16,000 g for 20 min at 4℃. The protein concentration was determined using the Bradford method (Bio-Rad, Hercules, CA, USA) according to the manufacturer's protocol. The two-dimensional (2D) gel electrophoresis separated the proteins. First dimensional isoelectric focusing was performed on an Ettan IPGphor System by using immobilized pH gradients (IPG, pH 4-7, 17cm, Bio-Rad) gel strips. The gel strips were rehydrated in sample buffer (8 M urea, 2% CHAPS, 20 mM DTT, 0.5% IPG buffer, bromophenol blue) that contained sample proteins at 30 V for 13 h at 20℃. IEF was carried out as follows: 250 V for 15 min, 10000 V for 3 h, and then 10000 V to 50000 V. The gradient gels (7.5-17.5%) were prepared for SDS gel electrophoresis. The strips were equilibrated and electrophoresesd using Protein-XI electrophoresis equipment (Bio-Rad) at 10℃. The loading conditions were 5 mA per gel for 2 h and followed by 10 mA per gel for 10 h. The gels were fixed in a solution (12% acetic acid, 50% methanol) for 2 h, washed in 50% ethanol for 20 min, and then treated in 0.2% sodium thiosulfate for 1 min. The gels were washed with deionized water, impregnated in a silver solution (0.2% silver nitrate, 0.75 mL/L formaldehyde) for 20 min. The gel was developed in a solution (0.2% sodium carbonate, 0.5 mL/L formaldehyde) and the reaction was stopped by adding 1% acetic acid. The silver stained gels were scanned using Agfar ARCUS 1200™ (Agfar-Gevaert, Mortsel, Belgium). The intensity of protein spots in scanned gel images were analyzed using a standard protocol for PDQuest software (Bio-Rad). The protein spots were were excised and prepared for mass spectrometry (MS) after the process of destaining, reduction, alkylation, and tryptic digestion. The extracted peptides were analyzed in a Voyager-DE™ STR biospectrometry workstation (Applied Biosystem, Forster city, CA, USA) for MALDI-TOF mass spectrometric analysis. Proteins were identified using search programs MS-Fit and ProFound. SWISS-PROT and NCBI were used as the protein sequence databases.
Total RNA of right cerebral cortex was isolated using Trizol reagent according to the manufacturer's instruction (Invitrogen, Carlsbad, CA, USA). Total RNA (1 µg) from each sample was reverse-transcribed into complementary DNA with superscript first-strand system for RT-PCR (Invitrogen) based on the manufacturer's protocol. The following primers were used: hippocalcin primer (207 bp), forward primer: 5'-ACGCCAACTTCTTCCCCTATG-3', reverse primer; 5'-AGCCATCAGCGTCTTTGTTT-3'; actin primer (238 bp), forward primer: 5'-GGGTCAGAAGGACTCCTACG-3', reverse primer; 5'-TTTCACTGCGGCTGATGTAG-3'. The PCR reaction was carried out as followed: 5 min at 94℃, 30 sec at 94℃, 30 sec at 54℃, 1 min at 72℃ and 10 min at 72℃. The samples were amplified 30 cycles. PCR products were electrophoresed on a 1% agarose gel was and visualized under UV light.
Western blot analysis was performed as previously described [15]. Briefly, the right cerebral cortex were homogenized and lysed in buffer [1% Triton X-100, 1 mM EDTA in 1×PBS (pH 7.4)] containing 200 µM phenylmethylsulfonyl fluoride and 10 µM leupeptin. The lysates were centrifuged at 12,000 rpm for 20 min at 4℃. The protein concentration of each lysate was determined using the bicinchoninic acid (BCA) kit (Pierce, Rockford, IL, USA). Protein samples (30 µg) were applied to each lane on 10% SDS-polyacrylamide gel electrophoresis. The gels were electrophoresed, and then transferred to a poly-vinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA) and membranes were washed in Tris-buffered saline containing 0.1% Tween-20 (TBST). And then membranes were incubated with the following antibodies: anti-hippocalcin (diluted 1:1000, Cell Signaling Technology, Beverly, MA, USA), and anti-actin (diluted 1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA). The membrane was incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (1:1,000, Cell Signaling Technology) for hippocalcin and HRP-conjugated anti-mouse IgG for actin. All visualization of immunoreactive bands was performed using ECL Western blot analysis system (Amersham Pharmacia Biotech, Piscataway, NJ, USA).
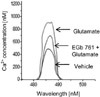
The mouse hippocampal cell lines, HT22 were cultured in Dulbecco's Eagle's medium (DMEM, without L-glutamine) supplemented with 10% fetal bovine serum as previously described method [16]. HT22 cells were seeded on 60-mm culture dishes at 100,000 cells per dish. The medium was changed and cells were treated with 5 mM glutamate (Sigma) or vehicle for 24 h. EGb 761 (5 mM) was treated 1 h before the addition of glutamate. The intracellular Ca2+ concentration was analyzed using calcium indicator acetoxymethylester fura-2 as previously described manual [17]. The cells were incubated in the DMEM media containing 10 µM fura-2/AM at 37℃ for 1 h. The cells were collected and washed with Locke's solution (pH 7.3) and then centrifuged at 1000 g for 5 min. Fura-2 fluorescence was monitored by luminescence spectrophotometer (LS50B, Perkin Elmer, Boston, MA, USA). The cells were alternatively excited from Xenon lamp by 340 and 380 nm wavelength, and the emission fluorescence intensity at 510 nm was revealed by a photon-counting photomultiplier. Fluorescence signals were measured using a MicroVax II computer and software (Origin 7). Calcium concentration was analyzed using the ratio method as previously mentioned equation [18]. [Ca2+]i=Kd(RRmin)/(RmaxR) xSf2/Sb2. Kd is the dissociation constant of fura-2 for Ca2+ and is assumed to be 224 nmol/L in cytosolic condition. R is the fluorescence ratio at 340 and 380 nm. Rmax is the ratio with saturating Ca2+, Rmin is the ratio with zero Ca2+. Sf2 is fluorescence at 380 nm in the absence of Ca2+ and Sb2 is fluorescence value at 380 nm with saturating Ca2+.
All data are expressed as mean±SEM. The intensity analysis was carried out using SigmaGel 1.0 (Jandel Scientific, San Rafael, CA, USA) and SigmaPlot 4.0 (SPSS Inc., Point Richmond, CA, USA). The results in each group were compared by one-way analysis of variance (ANOVA) followed by Student's t-test. The difference for comparison was considered significant at *P<0.05.
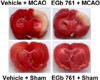
In supplement figure, we showed that the neuroprotective effect of EGb 761 against MCAO injury using TTC staining. EGb 761 administration decreases infarct volume, compared with the MCAO-operated without ferulic acid. Using a proteomic approach, hippocalcin protein spots were differentially expressed in the cerebral cortices of vehicle+MCAO and EGb 761+MCAO treated animals (Figure 1). The peptide mass of hippocalcin is 10/102 and the sequence of this protein is 52%. Hippocalcin expression was decreased in vehicle-treated animals, whereas the decrease in hippocalcin expression was prevented in EGb 761+MCAO animals. Hippocalcin levels were 0.63±0.02 and 0.91±0.02 in vehicle- and EGb 761-treated animals during MCAO, respectively. RT-PCR and Western blot analyses showed changes of hippocalcin levels in vehicle- and EGb 761-treated animals during MCAO. Transcript levels of hippocalcin were 0.42±0.02 and 0.87±0.01 in vehicle+MCAO and EGb 761+MCAO, respectively (Figure 2). Hippocalcin protein levels were also decreased in vehicle+MCAO animals compared to sham-operated controls, whereas EGb 761 pretreatment prevented the injury-induced decrease in hippocalcin protein levels. Hippocalcin protein levels were 0.31±0.03 and 0.93±0.02 in vehicle+MCAO and EGb 761+MCAO animals, respectively (Figure 3). We observed the relationship between intracellular Ca2+ levels and EGb 761 treatment in cultured hippocampal cells. Glutamate exposure increased intracellular Ca2+ levels, whereas EGb 761 treatment prevented the increase of intracellular Ca2+ levels (Figure 4).
EGb 761 has anti-oxidant and neuroprotective properties against neurological disorders and reduces neuronal cell death in global and focal brain ischemia [19,20]. We previously demonstrated the neuroprotective effect of EGb 761 against MCAO-induced cerebral ischemia [15,21]. The neuroprotective mechanism of EGb 761 in brain ischemic injury is very complex and is not clearly known. We previously observed that EGb 761 protects neuronal cells following focal cerebral ischemic injury by regulating of parvalbumin [22]. It is accepted that parvalbumin is a calcium buffering protein that plays a neuroprotective function by modulating intracellular calcium [23]. The present study additionally demonstrated the decrease of hippocalcin expression during MCAO. A proteomic approach elucidated that EGb 761 attenuates the MCAO injury-induced decrease in hippocalcin expression during MCAO. Hippocalcin acts as an endogenous diffusible calcium buffering protein and maintenances Ca2+ homeostasis in the central nervous system by binding to Ca2+ in the cytoplasm [7-10]. Calcium overload in neurons induces apoptotic cell death and leads to central nervous system disorders [11]. Therefore, we hypothesize that hippocalcin contributes to the neuroprotective mechanism. The results of this study provide evidence regarding the role of EGb 761 in neuroprotective mechanisms. The maintenance of hippocalcin levels in brain ischemia may inhibit the overload of calcium induced by ischemia and protect neurons against ischemic injury.
Ischemic injury-induced hypoxia triggers the activation of phospholipases and proteases. The activation of these enzymes leads to cell membrane damage, induces the elevation of intracellular calcium levels, and consequently disrupts cell calcium homeostasis [24-27]. Thus, calcium buffering proteins play critical roles in the determination of neuronal cell destiny [8]. Our results suggest that cerebral ischemic injury decreases hippocalcin expression, whereas EGb 761 pretreatment maintains hippocalcin expression at levels similar to those of sham-operated animals. In this study, we demonstrate that EGb 761 regulates hippocalcin expression in ischemic brain injury and consequently modulates intracellular calcium levels during cerebral ischemia. This study showed the regulation of intracellular calcium levels by EGb 761 in glutamate-induced neuronal cells injury. It is accepted that the increase of intracellular calcium induces cellular degradation and death [24]. The glutamate exposure increases the intracellular calcium concentration and EGb 761 attenuates the glutamate exposure-induced increase in intracellular calcium levels. Hiipocalcin is a critical protein that controls calcium homeostasis in the central nervous system. In a MCAO animal model, EGb 761 prevents decreases of hippocalcin in MCAO-induced injury. Moreover, hippocalcin performs a neuroprotective function through the regulation of intracellular calcium. Although further studies are needed to elucidate the relationship between EGb 761 and hippocalcin, the present results demonstrate that EGb 761 may maintain levels of hippocalcin expression and calcium homeostasis, thereby preventing neuronal cell death. In conclusion, our findings suggest that EGb 761 prevents neuronal cell death by regulating the calcium buffer protein, hippocalcin.



 XML Download
XML Download