

 PDF
PDF ePub
ePub Citation
Citation Print
Print


The pancreas is an important organ for regulating glucose metabolism in the body [1]. Glucose transported into β-cells induced by the increase of plasma glucose levels leads to insulin exocytosis, which manipulates the glucose levels [2]. Hyperglycemia has been known to be a primary factor in triggering diabetes in digestive organs [3]. Glutamate is the most important excitatory amino acid in the nervous system [4]. Recently, it has been reported that glutamate regulates the diverse function of digestive organs [5]. Several lines of evidence suggest that glutamate induces metabolic syndrome such as obesity and diabetes-like hyperglycemia [6,7]. Di Cairano et al [8] also reported that the glutamate transporter is expressed in pancreatic β-cells and is responsible for the protection of glutamate-induced β-cell death. These reports suggest that there may be an interaction between hyperglycemia and glutamate in the pancreas, since the pancreas plays a pivotal role in the regulation of glucose metabolism. However, the regulation of glutamate uptake by hyperglycemia in the pancreas has not been elucidated.
Diverse signaling molecules are involved in the onset of diabetes mellitus [9]. Among them, oxidative stress, which is manifested as increased levels of reactive oxygen species (ROS), has been generally reported to be associated with the development of diabetes [10,11]. Several lines of evidence have suggested that N-acetyl-L-cysteine (NAC) and quercetin protects the diabetes-related oxidative stress in the pancreas [12,13]. These reports suggest the possibility that oxidative stress is involved in the high glucose-induced regulation of glutamate uptake. Thus, this study was conducted to investigate the effect of high glucose on glutamate uptake and its relationship with oxidative stress in pancreatic β-cells.
Materials and Methods
Materials
RPMI 1640medium, fetal bovine serum (FBS), penicillin, and streptomycin were obtained from Hyclone (Logan, UT, USA). HIT-T15 cells were obtained from the American-Type Culture Collection (ATCC, Manassas, VA, USA). D-Glucose was obtained from Sigma (St. Louis, MO, USA). [3H]-L-glutamate was purchased from Dupont/NEN (Boston, MA, USA). All reagents were of the highest purity commercially available.
Cell cultures
We used HIT-T15 cells as a model to study pancreatic β-cells. HIT-T15 cells were cultured in RPMI 1640 medium containing 10% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37℃ in 5% CO2. Cells (1×104 cells/well) were seeded in a 96-well plate and pre-incubated with the same media in a serum-free condition for 24 h.
Cell viability assay
Cell viability was evaluated by the colorimetric MTT assay, using the CellTiter 96® AQueous One solution according to the manufacturer's instructions. Briey, cells (1×106 cells/well) were seeded in a 96-well plate with RPMI 1640 medium containing 10% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37℃ in 5% CO2. Then, the cells were pre-incubated with the same media in a serum-free condition (without FBS) for 24 h and were treated with 25 mM glucose in each experimental condition.
D-[2,3-3H]- Aspartate uptake
The D-[2,3-3H]-aspartate uptake experiments were carried out using a modification of the method described by Wu et al [14]. To study the D-[2,3-3H]-aspartate uptake, the culture medium was removed by aspiration, and the cells were gently washed twice with an uptake buffer (140 mM NaCl, 2 mM KCl, 1 mM KH2PO4, 10 mM MgCl2, 1 mM CaCl2, 5 mM glucose, 5 mM L-alanine, 5 µM indomethacin, and 10 mM HEPES/Tris, pH7.4). After washing, the cells were incubated in an uptake buffer containing 1 µCi/mL D-[2,3-3H]-aspartate at 37℃ for 1 h in 5% CO2 in a humidified cell culture incubator. At the end of the incubation period, the cells were washed three times with an ice-cold uptake buffer and then dissolved in 1 mL 0.1% sodium dodecyl sulfate (SDS). The level of D-[2,3-3H]-aspartate uptake incorporated intracellularly was determined by removing 900 µL of each sample and measuring the radioactivity in a LS 6500 liquid scintillation counter (Beckman Instruments, Fullerton, CA, USA). The remainder of each sample was used to determine the protein level by the method of Bradford [15]. The radioactivity counts in each sample were then normalized with respect to the protein and corrected for zero-time uptake per mg of protein. All the uptake measurements were carried out in triplicate.
Measurement of lipid peroxides
The levels of lipid peroxides (LPO) in the monolayer cells were determined by measuring the malondealdehyde content according to the method of Ohkawa et al [16]. ARPE-19 cells were exposed to 25 mM glucose for 24 h. After exposure terminated, the cells were harvested and sonicated. 100 µL of sonicated cells was mixed with 8% sodium dodecyl sulfate (100 µL), 0.8% 2-thiobarbituric acid (TBA, 200 µL) and 20% acetic acid (200 µL). The mixture was heated to 95 for 60 min. After reaction time, this mixture was cooled in ice-cold water. To extract the nonspecific red pigment, n-butanol-pyridine mixture (15:1 v/v, 1 mL) was added, and the mixture was shaken vigorously, and was then centrifuged at 4,000 rpm for 10 min. The upper organic layer was measured by spectrofluorometry at an emission wavelength of 553 nm with an excitation wavelength of 515 nm. 1,1,3,3-Tetraethoxy-propane was used as a standard and the values of LPO for samples were expressed as nmol/mg protein.
Results
Dose-dependent effect of high glucose on glutamate uptake
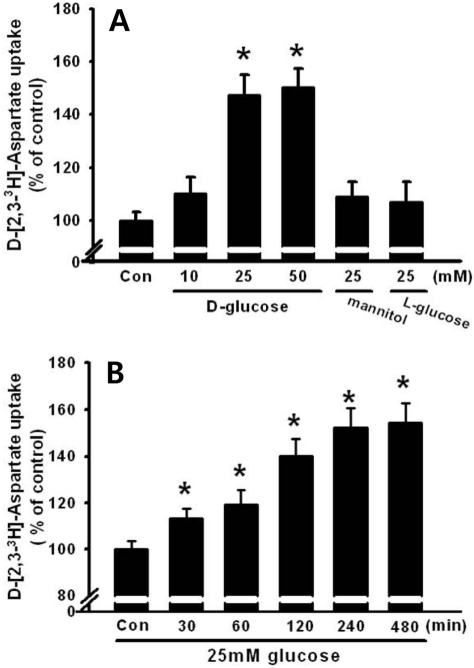
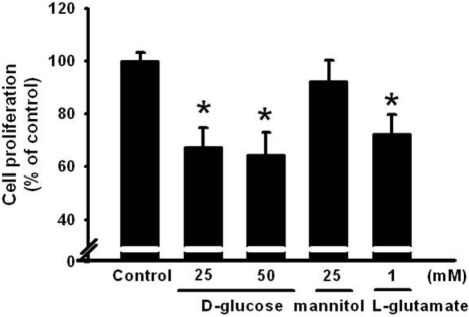
To examine the dose-dependent effect of high glucose on glutamate uptake, the HIT-15 cells were treated with different dosages of glucose (10, 25,or 50 mM), 25 mM mannitol, or 25 mM L-glucose. As shown in Figure 1A, 25 mM and 50 mM glucose, but not 10 mM glucose, significantly stimulated D-[2,3-3H]-aspartate uptake. However, treatment with 25 mM mannitol or L-glucose did not further change D-[2,3-3H]-aspartate uptake, suggesting that the uptake was glucose specific (Figure 1B). In addition, we examined the time dependent effect of glucose. Thus, HIT-15 cells were exposed to 25 mM glucose for 0-480 min. As shown in Figure 1B, 25 mM glucose significantly stimulated D-[2,3-3H]-aspartate uptake for over 30 min, with a maximum effect at 480 min after high glucose treatment. Thus, 25 mM glucose for 8 h was used in subsequent experiments. Furthermore, we examined high glucose and glutamate induced cell death in HIT-15 cells. Figure 2 demonstrates that high glucose (25 mM and 50 mM glucose), but not 25 mM mannitol, induced the decrease of cell viability. Similarly, the treatment of 1 mM glutamate also decreased the cell viability.
The involvement of oxidative stress in high glucose-induced stimulation of glutamate uptake and cell viability
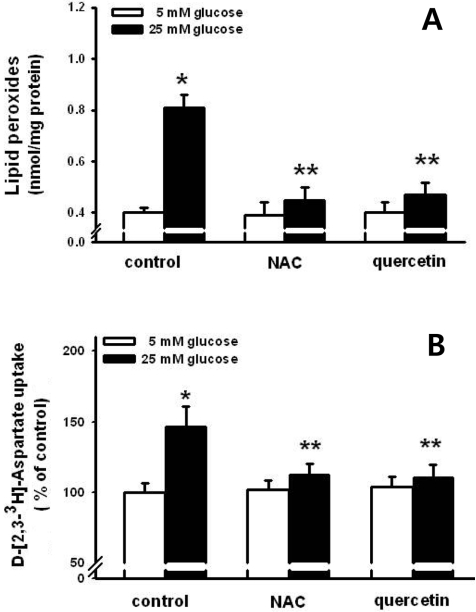
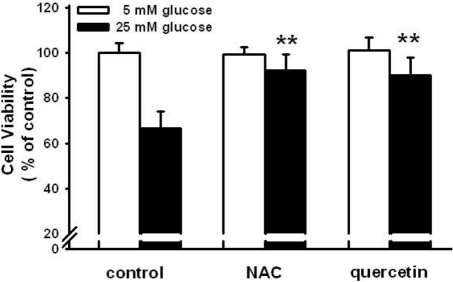
The relationship between oxidative stress and high glucose-induced stimulation in glutamate uptake was examined. NAC (1 mM) and quercetin (100 µM) antioxidants were used to treat the HIT-15 cells prior to incubation with 25 mM glucose. As shown in Figure 3A, 25 mM glucose induced the increase of LPO formation. NAC and quercetin both blocked high glucose-induced LPO formation. Figure 3B also demonstrated that NAC and quercetin blocked the high glucose-induced stimulation of glutamate uptake. NAC and quercetin prevented the high glucose-induced decrease of cell viability (Figure 4). These results suggest that oxidative stress is involved in the high glucose-induced stimulation of glutamate uptake and the decrease of cell viability.
Discussion
This study firstly demonstrated that high glucose stimulated the glutamate uptake in hamster pancreatic β-cells. Until now, the regulation of glutamate was restricted to the nervous system. However, several lines of evidence have suggested that the metabolism of glutamate is important in the function of non-neural tissues [17-19]. In the present study, high glucose stimulated the glutamate uptake in pancreatic β-cells. These stimulatory effects are D-glucose-specific and are unlikely due to an osmotic effect since the responses were not mimicked by mannitol or L-glucose. To our knowledge, this is the first report in which hyperglycemia stimulated the glutamate uptake in pancreatic β-cells. In pancreatic β-cells, glial glutamate transporter 1 and the vesicle glutamate transporter are expressed [20,21]. In the present study, we did not examine the isoforms of the glutamate transporters. This remains to be studied. Our result also demonstrated that high glucose and glutamate induced the decrease of cell viability. Our result is inconsistent with several reports. Gong et al [22] reported that hyperglycemia induces apoptosis of pancreatic islet endothelial cells. Venieratos et al [23] also showed that high glucose induces apoptosis in mouse pancreatic β-cells. Based upon our result and these reports, we provide the evidence that high glucose-induced stimulation of glutamate is associated with the apoptosis of pancreatic β-cells.
Changes in signal transduction pathways may potentially play an important role in the overall effect of glucose on glutamate uptake. ROS accumulation induces oxidative stress. Increased ROS occurs due to an imbalance of processes that produce ROS and processes that reduce ROS (antioxidant systems) in the pancreas [24]. In the present study, high glucose increased the LPO formation and antioxidants such as NAC and quercetin prevented the high glucose-induced stimulation of glutamate uptake and the decrease of cell viability. Zhang et al [25] also reported a similar result in which high glucose increased oxidative stress and these phenomena lead to β-cell apoptosis. However, they did not examine the role of glutamate. In agreement with our results, Penugonda et al [26] demonstrated that NAC derivative protects the glutamate-induced cytotoxicity in the neuronal cell line. High glucose-induced stimulation of glutamate uptake and stimulation of LPO formation have been reported in many various cells and tissues [27,28]. Nevertheless, there is still no direct evidence demonstrating the relationship between glutamate uptake and LPO formation in pancreatic β-cells. Here, we provide further evidence that high glucose-induced induced increase of LPO formation leads to the stimulation of glutamate uptake, which may be associated with the apoptosis of pancreatic β-cells. There is a possibility that the high glucose-induced stimulation of glutamate uptake triggers the increase of oxidative stress, since L glutamate enhances cell toxicity via the increase of oxidative stress [29]. It is speculated that mechanisms other than oxidative stress could have a role in the glucose-induced stimulation of glutamate uptake. This needs to be elucidated in a further study. In summary, in the HIT-T15 cells, the pancreatic β-cells, oxidative stress appears to mediate the high glucose-induced stimulation of glutamate uptake. In addition, oxidative stress is also involved in mediating the high glucose-induced decrease of cell viability. Therefore, our findings may provide new insights into the pathophysiological mechanisms of glutamate uptake in diabetes mellitus. In conclusion, high glucose induced the stimulation of glutamate uptake and decrease of cell viability via oxidative stress in the pancreatic β-cells.
 XML Download
XML Download