

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Reactive oxygen species (ROS) produced by excitotoxic injury during cerebral ischemia contribute to neuronal cell injury. Glutamate, an important excitatory neurotransmitter in the central nervous system, stimulates a Ca2+-dependent release of arachidonic acid from neuronal cells through the activation of N-methyl-D-aspartate (NMDA) receptors [1]. Almeida et al. [2] reported that glutamate neurotoxicity is a correlate of glutathione depletion. High potassium-induced glutamate release stimulates the production of arachidonic acid in cortical neurons [3]. Glutamate is also implicated in cognitive function, such as learning and memory and in neurotoxic insult [4]. The activation of NMDA receptors increases arachidonic acid release in cultured brain cells [5]. The activation of 5-lipoxygenase in arachidonic acid metabolism appears to correlate with neuronal death caused by deprivation of endogenous glutathione (GSH) [6]. Although many modulators or blockers that act on NMDA receptors have been developed, almost all clinical trials have failed. Thus, it is urgent to develop a suitable neuroprotective compound for the rescue of neuronal cells during ischemic insults in brain. Esculetin is a coumarin derivative that exhibits various pharmacological actions: it has antioxidant, anti-inflammatory, and anti-edematous effects [7]. L-Buthionine-(S,R)-sulfoximine (BSO), a glutathione synthesis inhibitor, exacerbates glutamate- or NMDA-induced neurotoxicity in cortical cultures [8]. Increases in the release of reduced glutathione in cerebral ischemia has been shown to exacerbate the excitotoxic insult via NMDA receptors [9,10]. In this study, we examined whether esculetin, a lipoxygenase inhibitor, can rescue neuronal cells via modulatory actions on NMDA receptors, and whether, in primary cortical cultures, its protective actions can be ascribed to regulation of glutathione metabolism, including antioxidant effects.
Materials and Methods
Mixed cortical cell cultures containing both glia and neurons, were prepared from ICR mouse brains at 15-16 days of gestation. Briefly, dissociated neocortical cells (3.5×105 cells/well) were plated onto primaria-coated 24-well plates (Falcon) containing a glial bed in plating medium consisting of Eagle's minimal essential medium (MEM; Earle's salts, supplied glutamine-free) supplemented with 20 mM glucose, 2 mM L-glutamine, 5% fetal bovine serum, and 5% horse serum. Cytosine arabinoside (10 µM) was added 5 days after plating to halt the growth of non-neuronal cells.
Cultures were maintained at 37℃ in a humidified CO2 incubator and used for experiments between days 12 and 14 in vitro. Glial cultures were prepared from postnatal mice (1-3 days) and plated at 1×105 cells/well in plating medium supplemented with 10% horse serum, 10% fetal bovine serum, and 10 ng/mL epidermal growth factor. After 2 weeks in vitro, cytosine arabinoside was added to the cultures, which were fed weekly with the same medium with 10% horse serum used for mixed cultures. We added glycine (10 µM final concentration) to all media. We pre-incubated cultures with esculetin for 2 h before inducing neurotoxicity.
For measuring 45Ca2+ uptake, cultured cells were washed with HEPES-buffered control salt solution (HCSS), and then incubated with NMDA (150 µM) in the presence of MK-801 (an NMDA receptor inhibitor, 10 µM) or esculetin (10, 100 µM) in HCSS containing CaCl2 (final activity: 1.0 µCi/mL). After 5 min, exposure solution was washed away using four boluses of HCSS and cells were lysed by addition of 0.2% sodium dodecyl sulfate solution. Aliquots of lysed cells were added to scintillation counting solution for the counting of beta emissions [11].
For protein preparation, cells from three culture plates were pooled in 0.5 mL of 0.1 M phosphate buffer (pH 7.4) and homogenized. The homogenate was centrifuged for 30 min at 3,000 g at 4℃ and the supernatant, consisting of the cytosolic+mitochondrial fractions, was used in enzyme assays [12].
Glutathione peroxidase (GPx) activity was measured using an NADPH reduction assay following the technique of Lawrence and Burke [13]. Soluble cell proteins were added to a reaction mixture containing reduced glutathione, glutathione reductase, and NADPH in phosphate buffer. The reaction was initiated by adding H2O2, the absorbance decrease at 340 nm was recorded, and the activity in the absence of cellular protein was subtracted. Results were based on a molar extinction coefficient for NADPH of 6.22×103 M-1 cm-1. One unit of GPx is defined as µmoles NADPH oxidized/min, and results are expressed in µmoles/min/mg protein.
Glutathione reductase (GR) activity was measured using an NADPH reduction assay following the technique of Eklöw et al. [14] using oxidized glutathione disulfide (GSSG) as substrate. GR activity was monitored by decreases in NADPH absorbance at 340 nm at 37℃. Results were based on a molar extinction coefficient for NADPH of 6.22×103 M-1 cm-1. One unit of GR is defined as µmoles NADPH oxidized/min, and results are expressed in µmoles/min/mg protein. Protein content was measured by the method of Lowry et al [15] with bovine serum albumin as a standard.
Neuronal damage was assessed by measuring the amount of LDH released into the medium by damaged cells 24 h after NMDA or BSO treatment. Morphological confirmation was obtained by trypan blue staining. Data are expressed as the mean±standard error of the mean (SEM) and analyzed for statistical significance by one-way ANOVA using a post-hoc Student-Neuman-Keuls test for multiple comparisons.
Results
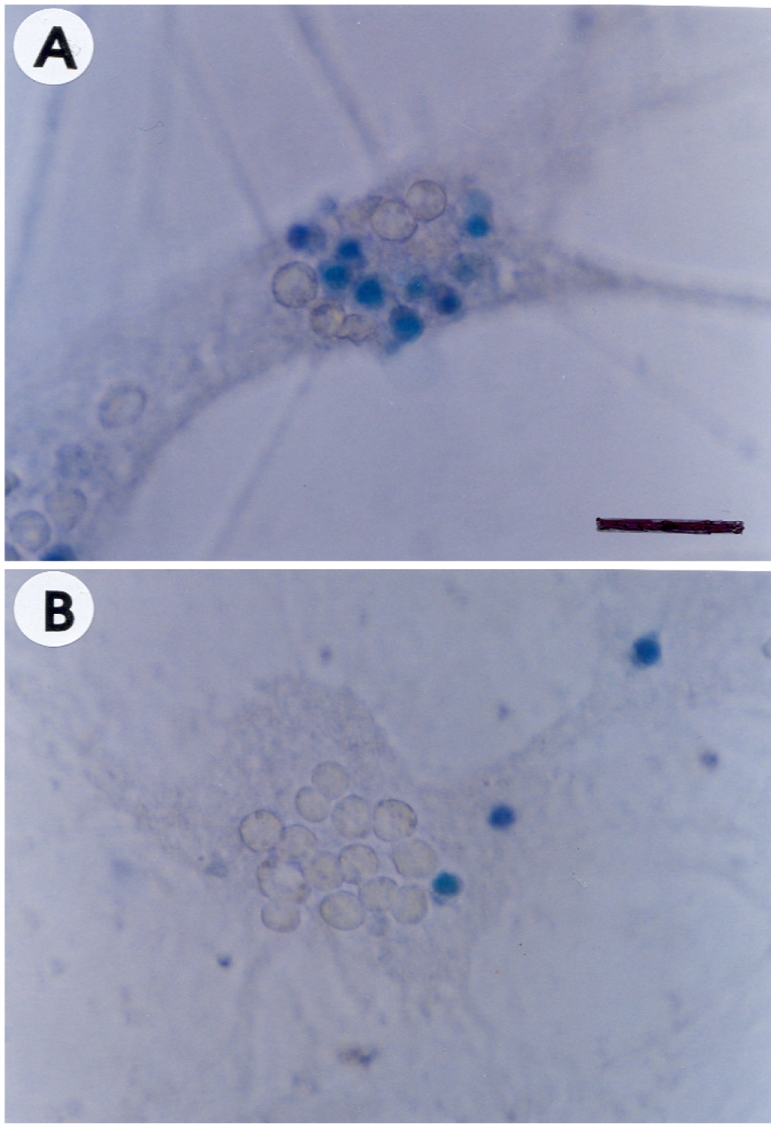
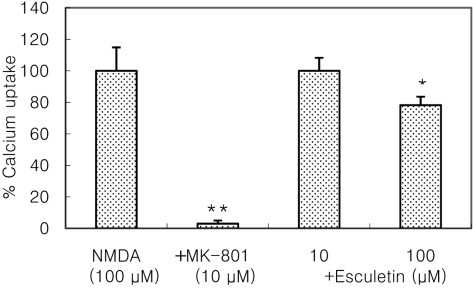
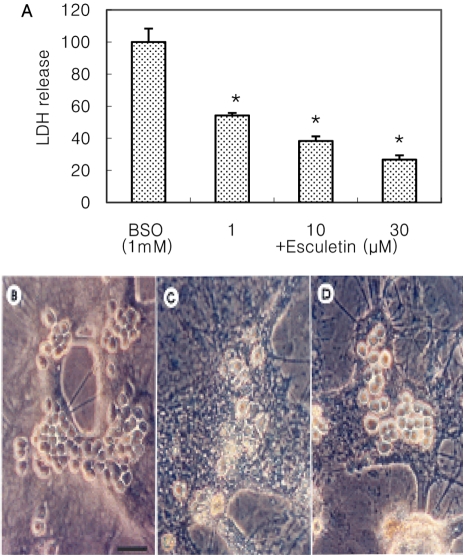
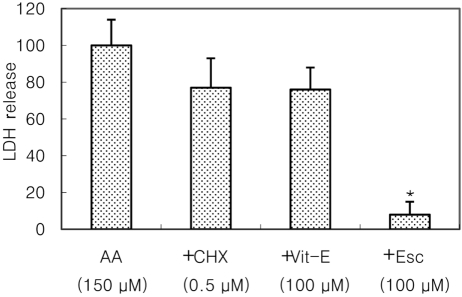
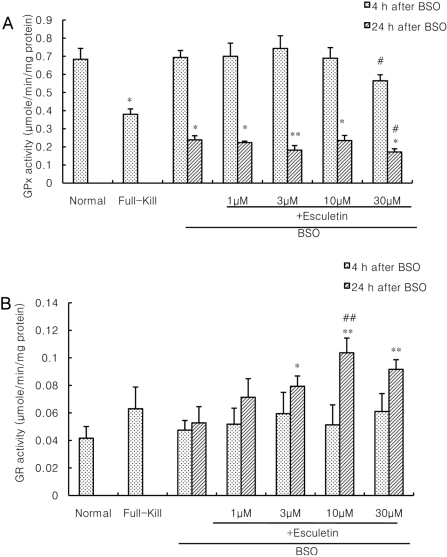
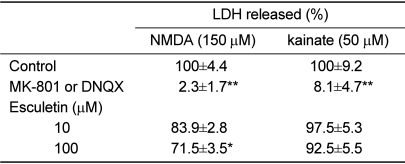
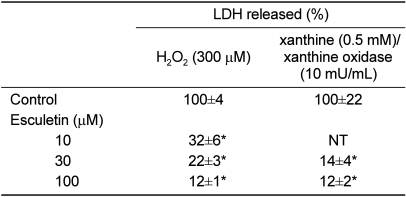
Cultured cortical cells exposed to 150 µM NMDA or 50 µM kainic acid (KA) generated moderate (50-70%) neuronal damage as indicated by increased LDH release into the bathing medium after 24 h. Pre-exposure of esculetin (10, 100 µM) for 2 h attenuated NMDA neurotoxicity by 16% and 29%, respectively, but did not show any inhibitory effect against KA neurotoxicity (Table 1). At the highest concentration, esculetin significantly ameliorated NMDA-induced neuronal injury. Esculetin dose-dependently and significantly inhibited the neurotoxicity induced by H2O2 or superoxide anions (O2·-) produced by xanthine/xanthine oxidase co-treatment (Table 2). Morphologically, NMDA-induced neurotoxicity manifested as loss of cell bodies (Figure 1A); esculetin rescued many neurons from NMDA neurotoxicity (Figure 1B). To determine whether esculetin modulates NMDA receptors, we tested the effects of NMDA alone or NMDA+esculetin. Uptake of 45Ca2+ increased after NMDA exposure for 5 min. MK-801, a non-competitive NMDA receptor antagonist, blocked 45Ca2+ uptake as well as LDH release. At 100 µM, esculetin significantly ameliorated the NMDA-induced rise in 45Ca2+ uptake (Figure 2). To investigate the interrelationship between lipoxygenase (LOX) inhibition and glutathione metabolism, we measured esculetin modifiable GSH depletion-induced neurotoxicity. After 2-h pretreatment with esculetin or vehicle, we exposed the cultured cells to 1 mM BSO for 24 h. Esculetin, a LOX inhibitor (1, 10 or 30 µM) significantly attenuated BSO-induced neuronal injury by 46, 62, and 73%, respectively (Figure 3A). We morphologically confirmed the protective effect of 30 µM esculetin (Figures 3B-3D). At 150 µM arachidonic acid, the neuronal cells were damaged 70-80%. Cycloheximide, an inhibitor of de novo protein synthesis, and vitamin E, an antioxidant, reduced neuronal damage by 25% (Figure 4). In contrast, esculetin (100 µM) significantly inhibited the neuronal injury by 90% (Figure 4). GPx activity was decreased by BSO treatment, but BSO did not change GR activity at 24 h compared to the normal group (Figures 5A and 5B). Here, esculetin at 3-30 µM concentration-dependently elevated GR activity compared to BSO alone (Figure 5B) but caused little change in GPx activity (Figure 5A). These findings suggest that esculetin inhibits oxidized glutathione (GSSG) accumulation by increasing GR activity, thus sparing the pool of reduced glutathione (GSH).
Discussion
Ischemic injury, especially NMDA neurotoxicity, is known to activate calcium-dependent lipoxygenase pathways [6]. So far, almost all NMDA antagonists have failed to protect neuronal cells from cerebral ischemia clinically due to their psychiatric side effects. In this study, esculetin, a non-competitive lipoxygenase inhibitor, ameliorated NMDA-induced neurotoxicity.
However, esculetin did not show protective effects against KA neurotoxicity. These results are consistent with our previous in vivo results [16]. In addition, we confirmed that the protective effect of esculetin against NMDA toxicity can be partly attributed to its ability to modulate NMDA receptors directly or indirectly, as demonstrated by the results for calcium uptake. When NMDA receptor overactivation is initiated by excitotoxic insults, release of arachidonic acid and increases in lipoxygenase activity can occur [6,17]. As in other reports, esculetin strongly inhibited arachidonic acid-induced neurotoxicity. The antioxidant vitamin E and the de novo protein synthesis inhibitor cycloheximide exhibited only weak protective effects. This means that esculetin may contribute to the diminution of neuronal injury that occurs in ischemic insults by attenuating the NMDA receptor-mediated arachidonic acid cascade. Interestingly, esculetin also had a sparing effect on the pool of GSH by halting GSH turnover to GSSG. Esculetin induced a powerful inhibition of NMDA-induced glutathione depletion. Furthermore, esculetin may contribute to the rescue of neuronal cells from NMDA neurotoxicity by scavenging H2O2 or O2·-
We conclude that esculetin is a potential neuroprotectant in cerebral ischemia that appears to works by modulating NMDA receptors and the metabolism of arachidonic acid or glutathione.
 XML Download
XML Download