

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Mycoplasmas belong to the class Mollicutes and are among the smallest free-living microorganisms capable of auto-replication. Mycoplasmas are highly fastidious bacteria, difficult to culture and slow growing [1]. Many species are important veterinary pathogens causing respiratory infection, mastitis, conjunctivitis, arthritis, and occasionally abortion [1]. Also, in the field of laboratory animal medicine, it has been reported that mycoplasma infections are very common and considered highly contagious [2]. Major mycoplasmas of laboratory animals include Mycoplasma (M.) hyopneumoniae, M. pulmonis, M. collies, M. neurolyticum, M. arthritiditis, M. hyorhinis, and M. mycoides [3]. Identification of mycoplasmas as the causative agents of disease is often hindered by the lack of a rapid diagnostic test together with similarities in the clinical diseases that they cause. Conventional methods of diagnosis are based on culture and serological tests, such as the complement fixation test [4], enzyme-linked immunosorbent assays [5], and immunoblotting [6], and can be time-consuming, insensitive, and nonspecific. It is necessary to clarify the current status of mycoplasma contamination in laboratory animal colonies because mycoplasmas are prevalent in animals in commercial and research facilities [7].
Mycoplsama infection in laboratory animals interferes with biomedical research [8]. The transmission of M. pulmonis by aerosol from an infected to a healthy rat by sneezing at a distance of about 120 cm strongly suggests that such transfers may happen between rats and humans [2]. The technicians cleaning cages of facilities with rats infected with M. pulmonis are liable to be infected with these bacteria. Although mycoplasmas show specificity for their hosts, isolation of some mycoplasmas from unusual hosts has already been reported [9,10].
Polymerase chain reaction (PCR) provides a powerful technique for identifying different mycoplasmas and studying homology between their nucleic acids. However, those kinds of PCR assays require multiple assays because there are a lot of mycoplasma species [1]. Also, PCR has the high risk of the false-positive reaction by contamination and of the false-negative result by enzymatic inhibitors [11-13]. In order to avoid problems related to nucleic acid amplification, efforts have been made to develop specific hybridization assays such as dot blot hybridization (DBH) and in situ hybridization [14]. DBH is a simple and specific method for detection of pathogens and has been reported to be a method with higher specificity and lower sensitivity compared to PCR assays [15-17].
The aims of the present study were to develop a mycoplasma genus-specific PCR/DBH assay using a 16S ribosomal DNA gene. Such an assay might be a fast and practical method for detection and identification of Mycoplsama species.
Material and Methods
Microorganisms and growth conditions
M. hyopneumoniae (ATCC 25934) was obtained from the American Type Culture Collection (ATCC, Rockville, USA). The mycoplasmas were grown in modified Friis medium [18], containing 20% porcine serum (Gibco BRL, Rockville, USA), 5% fresh yeast extract (Gibco-BRL), methicillin (0.15 mg/mL; Sigma-Aldrich Canada, Oakville, Canada), bacitracin (0.15 mg/mL; Sigma-Aldrich Canada), and thallium acetate (0.08 mg/mL; Sigma-Aldrich). The cells were harvested by centrifugation at 12,000 g for 30 min at 4℃, washed three times, and suspended in 0.1M phosphate-buffered saline (PBS), pH 7.4.
Polymerase chain reaction
M. hyopneumoniae DNA was extracted from cultured M. hyopneumoniae using an AccuPrep Genomic DNA extraction kit (Bioneer, Daejeon, Korea) according to the manufacturer's instructions. The DNA was eluted in Tris-EDTA buffer (pH 8.0), and an aliquot was used for the PCR amplification. Amplification of the V3 region of the 16S ribosomal DNA was performed with consensus primers GC-341F (5'-CGC CCG CCG CGC GCG GCG GGC GGG GCG GGG GCA CGG GGG GCC TAC GGG AGG CAG CAG) and 534R (5'-ATT ACC GCG GCT GCT GG), which were based on the sequences reported by Weisburg et al. [19]. The template DNA (50 ng) and 20 pmol of each primer were added to a PCR mixture tube (AccuPower PCR PreMix; Bioneer) containing 2.5 U of Taq DNA polymerase, 250 µM each deoxynucleoside triphosphate, 10 mM Tris-HCl (pH 8.3), 40 mM KCl, 1.5 mM MgCl2, and the gel loading dye. The volume was adjusted with distilled water to 20 µL. The reaction mixture was subjected to denaturation at 94℃ for 5 min followed by 30 cycles of 95℃ for 1 min, 55℃ for 45 sec, and 72℃ for 1 min, and then a final extension step of 72℃ for 10 min was done. Samples were kept at 4℃ until analysis. Reactions were conducted using a My Genie 32 Thermal Block PCR (Bioneer).
Preparation of non-radioactive mycoplasma consensus DNA probe
The mycoplasma consensus DNA probes were constructed by PCR and labeled with digoxigenin after the amplification reaction. The mycoplasma consensus PCR using 16S ribosomal DNA pairs, was performed as described previously [3]. After amplification, PCR products were purified using Wizard PCR preps (Promega Biotech, Medison, USA). Purified PCR products were labeled by random priming with digoxigenin-dUTP (Roche Applied Science, Mannheim, Germany) or by means of a commercial kit according to the manufacturer's instructions.
PCR/dot blot hybridization
The detection of PCR products using a non-radioactive DNA probe was conducted by PCR/DBH assay to allow for sensitive and specific detection of target DNA. Dot blotting was achieved by direct application on a positively charged nylon membrane (Roche Applied Science). The PCR products after primary amplification were dotted on the nylon membrane. The membrane was immersed in 0.4M NaOH for 5 min and then in neutralizing buffer for 5 min. After rinsing in 2× saline-sodium citrate buffer (SSC), cross-linking between the applied DNA and the membranes was done using UV cross-linker (Stratagene, La Jolla, CA, USA). Hybridization solutions consisted of 5×SSC, 2% buffered blocking solution (Roche Applied Science), 0.1% N-lauroylsarcosine, and 0.02% sodium dodecyl sulfate. Digoxigenin (DIG)-labeled probe, which was denatured by boiling for 10 min and chilling in ice, was added in hybridization solution at 0.1 mg/mL before the hybridization. After pre-hybridization at 50℃ for 1 hour, the membrane was hybridized at 50℃ for 3 h and washed with 1×SSC at 60℃ for 10 min and 1× washing buffer (Roche Applied Science) each. For detection of hybridization, the membrane was incubated with anti-digoxigenin conjugated with alkaline phosphatase (Roche Applied Science) and then colorized with nitroblue tetrazolium (NBT) and 5-bromocresyl-3-indolyl-phosphate (BCIP). Thereafter, the development of a dark purple positive reaction was allowed to proceed for 10-30 min in the dark.
Sensitivity and specificity
The sensitivity and specificity of PCR/DBH was evaluated. Purified M. hyopneumoniae DNA samples ranging from 107 to 101 pg were used for the primary target amplification. After amplification of those template DNA samples, PCR/DBH was done with those primary amplicons. To identify the sensitivity of primary PCR amplification alone, ten microliters of the amplified PCR products were analyzed by 2% agarose gel electrophoresis and stained with ethidium bromide for product visualization. Also, to identify the sensitivity of DBH alone, purified M. hyopneumoniae DNA samples ranging from 107 to 101 pg were directly applied on positively charged nylon membranes (Roche Applied Science) and then DBH was done. The specificity of PCR/DBH was evaluated using template DNA samples such as Actinobacillus pleuropneumoniae and Pasteurella multocida, DNAs that were provided by professor C. Chae at Seoul National University in Korea.
Results
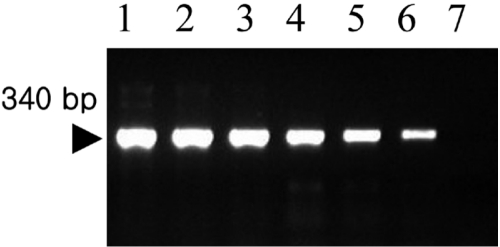
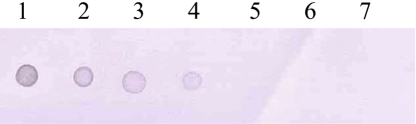
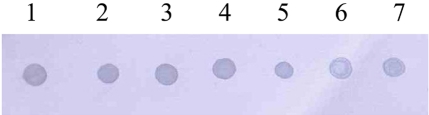
Amplification of the V3 region of the 16S ribosomal DNA with mycoplasma consensus primers GC-341F and 534R was performed for primary amplification of target DNA. In this study, Mycoplasma DNA was detected template DNA down to 102 pg (Figure 1). Because the samples were DNAs from cultured M. hyopneumoniae, control DNA was not need for the PCR reaction. The 16S rDNA gene (340 bp) was specifically amplified by PCR with the mycoplasma genus-specific primers (GC-341F and 534R). The strongest signal was shown in the PCR product of template DNA with 107 pg (Figure 1). The sensitivity of DBH alone was low (Figure 2). The detection limit for DBH was 104 pg mycoplasma DNAs. The highest intensity was observed in the signal of the sample DNA with 107 pg. As we decreased the amount of sample DNAs, the signal intensity became weak. A sample DNA with 103 pg did not generate any positive signals (Figure 2). PCR/DBH had higher sensitivity than PCR or DBH alone. The increased sensitivity of this technique was apparent even when sample DNA of 10 pg was detected as the positive signal and the intensities of each sample was higher than an equivalent sample that had undergone DBH alone (Figure 3). The specificity of PCR/DBH was confirmed using other bacterial DNAs with high homology in their sequences. No positive signals were observed in template DNA samples of Actinobacillus pleuropneumoniae and Pasteurella multocida in the PCR/DBH assay. However, PCR/DBH using Mycoplasma template DNA resulted in a strong positive signal (Figure 4).
Discussion
The diagnosis of mycoplasma infection is usually done by cultivation of the organism or by immunofluorescence tests performed on frozen thin lung sections with polyclonal antibodies [20,21]. However, due to the fastidious nature of mycoplasma species, their culture and serological identification may take up to 1 month. Serological detection methods are further hampered by cross-reactions that have been reported between M. hyopneumoniae, M. hyorhinis, and M. flocculare [22,23]. With the advances made in molecular biology during the last few years, more is known about mycoplasma species genes. Hence, other methods can be used as diagnostic tools for this organism. Recently, PCR methods were used to detect mycoplasma species [3]. These methods are rapid and sensitive, especially if nested PCR is used. However, PCR assays are subject to a high risk of contamination through DNA carry-over and may result frequently in false positive reactions [24-26]. Thus it is necessary to improve PCR sensitivity and therefore we developed a PCR/DBH assay. That assay is a very sensitive and specific tool for mycoplasma species detection. PCR sensitivity and specificity can be increased by hybridization methods of replicated DNA with specific labeled probe. In this study, we used non-radioactive labels as DBH probes, which have made these techniques more attractive for diagnostic laboratories, because they avoid problems relative to the short half-life of radioactive compounds, their disposal, and personnel safety [27,28]. Different methods have been developed for DNA probe labeling. In the past, DNA probes were labeled by radioactive isotopes (P32, I125) but diagnostics required and initiated development of rapid, non-radioactive methods for DNA probe labeling. For non-radioactive DNA probe labeling, different enzyme and immunological markers have been used [29] such as horseradish peroxidase (HRP), monoclonal and polyclonal antibodies to the DNA probe conjugated with alkaline phosphatase, and biotin conjugated DNA probes which, in with avidin-enzymatic complex produces a yellow color that can be measured by calorimetric or densitometric methods [30]. Recently, methods have been developed for non-radioactive labeling of DNA fragments by DIG-labeled dUTP randomly incorporated in the DNA. DNA probe built-in DIG-dUTP is detected by antidigoxigenin antibodies conjugated with alkaline phosphatase, which in reaction with X-phosphate and NBT salts produces a blue-stained line where the searched hybrid is positioned in the nitrocellulose membrane. DNA probe labeling was used in this study and the method was shown to be rapid, sensitive and specific, making it suitable for the detection of primary amplified Mycoplasma species DNA products. This allowed for increased sensitivity and specificity and quantification of mycoplasma species by DNA densitometry. The total assay time including the PCR procedure and DBH detection is 8 hours. The PCR/DBH assay that was established in this study is much more sensitive and specific compared with a one step PCR assay and DFA detection.
In conclusion, we developed a sensitive and reproducible method-mycoplasma consensus PCR/DBH- for the detection of mycoplasma species DNA. The PCR/DBH assay may be a valid tool for the diagnostic laboratory and can be an alternative to PCR assays for screening of a large number of samples to detect mycoplasma species.

 XML Download
XML Download