

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Characteristics of Skin Aging
Our society is entering an era of aging with increasing life expectancies owing to the remarkable advancement of medical science. An aging society means the ratio of elderly people over 65 years old comprises more than 7% of the overall population. Aging is an inevitable and natural process of life, but with increases in the aged population, social concerns of aging and anti-aging are greater than ever. All body organs show dysfunction and reduced capacity with aging (Kosmadaki and Gilchrest, 2004). In skin, aged phenotypes which are commonly characterized as increased wrinkling, sagging, and laxity are more apparent than in other internal organs. Since skin has a physiological function as a barrier for protection against environmental damages but also serves as a social interface between an individual and society (Robert et al, 2009), the interests and efforts for preventing skin aging are growing rapidly in the dermatology field and cosmetic industry.
Another feature of skin aging is that the skin suffers both intrinsic and extrinsic aging (Fisher et al, 2002) whereas internal body organs only undergo intrinsic aging. Intrinsic skin aging is an innate aging process resulted from the passage of time and is thus called chronological aging. It is generally determined genetically and has been thought to begin upon the onset of switch genes at a particular time according to a biologic clock programmed in the body (Uitto et al, 1989; Kosmadaki and Gilchrest, 2004). The tissue function of the skin deteriorates by intrinsic aging similar to internal body organs but the changes of cutaneous aging are easily recognized by clinical manifestations including fine wrinkling, loss of elasticity, and increased fragility (Gilchrest, 1982).
On the other hand, extrinsic skin aging is an environmentally-derived aging process. It can arise from external factors such as sun exposure, smoking (Turchin et al, 2009), nutrition, etc., of which UV irradiation from the sun damages human skin more severely than do other factors. It has been proposed that up to 90% of visible skin aging is due to cumulative exposure to sunlight (Sudel et al, 2005). Thus, extrinsic skin aging is termed photoaging (Kligman et al, 1989). Photoaged skin also reveals distinct deleterious changes such as coarse and deep wrinkling, thickened and leathery appearance, and irregular pigmentation (Kligman and Kligman, 1986). In general, photoaging is seen with intrinsic aging, especially in sun-exposed skin (Fisher et al, 2002). Whilst these intrinsic and extrinsic aging processes of skin have different etiologies and changes, they both exert harmful effects on dermal connective tissue (Chung et al, 2001; Fisher et al, 2002; Sardy, 2009). In this article, we summarize the molecular mechanisms of skin aging that have prominent effects on dermal matrix components and studies in in vivo mouse models.
Dermal Connective Tissue
Skin is made up with three cutaneous layers of epidermidis, dermis, and subcutaneous tissue. The epidermis is the most superficial layer and its main function is to protect the body from harmful environmental stimuli and diminish fluid loss. It is a multilayered stratified squamous epithelium which is constantly renewed throughout life by the keratinization, a process of shedding of cornified cells. Keratinocytes are the principal cells of the epidermis and maintain epidermal homeostasis (Blumenberg and Tomic-Canic, 1997). The dermis is located under the epidermis and is attached to it by the dermo-epidermal junction. Dermal connective tissue forms the dermis and contains extracellular matrix proteins such as collagen, elastic fibers, fibronectin, glycosaminoglycans, and proteoglycans which are produced and secreted into the extracelluar space by fibroblasts, the main cell type of the dermis (Makrantonaki and Zouboulis, 2007). Collagen is the most abundant protein in dermal connective tissue, making up to 70% of the dry skin mass (Gniadecka et al, 1998), and has a characteristic triple-helix configuration through an enzymatic process. The triple-helix complexes associate with small proteoglycans to form regularly arranged fibrillar structures called collagen bundles (Bateman and Chothia, 1996) that are responsible for conferring strength and support to human skin (Oxlund and Andreassen, 1980; Uitto, 1986). Elastic fibers are mainly composed of elastin and fibrillin, and are formed in a way in which cross-linked elastin forms the central fiber core and fibrillin-rich microfibrils surround the core. These account for less than 1-2% of the dermal weight but play a functional role in resisting deformational forces and providing elasticity by forming a fine network (Makrantonaki and Zouboulis, 2007). Proteoglycans are also non-collageneous proteins similar to elastic fibers comprising only 0.1-0.3% of the dry weight of skin, and fill the majority of extracellular interstitial spaces within the tissue (Frantz et al, 2010). They are responsible for the assembly of the extracellular matrix by affecting collagen fibrillogenesis and regulating the diameter and structure of collagen fibrils, thus preventing abnormal fibril assembly (Okamoto and Fujiwara, 2006). Decorin and dermatopontin are the main molecules of proteoglycans (Okamoto and Fujiwara, 2006). These various extracellular matrix proteins in the dermal connective tissue mainly contribute to the maintenance of skin integrity and architecture. Therefore, damages to the dermal connective tissue by the aging process can be closely related to structural changes of the skin like wrinkling, loss of elasticity, and sagging.
Molecular Mechanisms of Skin Aging
Cellular senescence of fibroblasts
Cellular senescence of dermis-derived cells that are maintaining homeostasis of the dermal connective tissue is a fundamental cause of skin aging and brings about growth retardation due to a decreased proliferative capacity of cells mostly by the intrinsic aging process. As in all organs, dermal fibroblasts responsible for producing matrix proteins also show replicative senescence with age (Praeger, 1986; Cristofalo and Pignolo, 1993). In the estimation of cellular senescence of human dermal fibroblasts, the population doubling-time of older cells (passage 28) was three times longer than of younger cells (passage 4) (Yoon et al, 2004). From the cell cycle profile by flow cytometry in this study, it was also found that older cells have increased G0/G1 phase cell populations and a decrease in the S and G2/M fractions. Growth reduction and cell cycle arrest at the G0/G1 phase can be ascribed to the repression of several regulatory genes that are important for cell cycle progression and cell proliferation (Yaar et al, 2002). These genes include c-fos that encodes a component of the AP-1 transcription factor (Seshadri and Campisi, 1990; Sheerin et al, 2001), Id1H and Id2H encoding helix-loop-helix proteins (Hara et al, 1994), the E2F family of transcription factors that induce the expression of the genes required for cell proliferation (Dimri et al, 1994), and CENP-F (mitosin) and KIF4FA which are related with the onset of mitosis and mitotic chromosomal positioning (Rattner et al, 1993; Lee and Kim, 2003); all of these are downregulated in senescent fibroblasts. On the contrary, the p21 and p16 inhibitors of cyclin-dependent protein kinases are up-regulated to interrupt the cell cycle progression (Noda et al, 1994; Hara et al, 1996). Many specific genes associated with cellular senescence have been identified but the existence of a master switch that induces the activity of these genes at a particular time remains to be determined (Jenkins, 2002).
Another mechanism for explaining cellular senescence is that age-associated changes in gene expression patterns and cellular proliferative capacity are likely to be under the control of telomeres (Kosmadaki and Gilchrest, 2004). Telomeres are the ends of linear chromosomes consisting of tandem repetitive DNA sequences. The length of telomeres decreases by up to 150 base pairs with cell division (Harley et al, 1990) and is inversely related to an individual's age (Allsopp et al, 1992). When telomeres become critically short, cells enter proliferative senescence and thus telomeres may serve as a biologic clock indicating whether cells are young or old (Wright and Shay, 1992). Therefore, telomere shortening can account for cellular senescence being mainly induced by intrinsic aging because it arises from serial cell division. However, it has also been determined that photoaging appears to influence telomere attrition (Li et al, 2003).
Decreased synthesis of matrix proteins
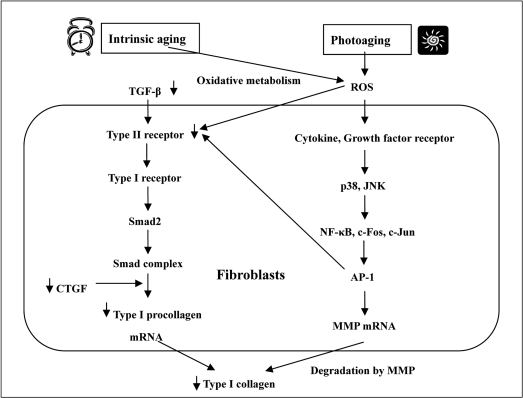
As a result of cellular senescence, non-dividing aged dermal fibroblasts accumulate in the dermal connective tissue and the synthesis of various extracellular matrix proteins by senescent fibroblasts is undoubtedly reduced. It is well known that the synthesis of fibrillar collagen, which is the major extracellular matrix protein and provides a supportive extracellular framework, is reduced in aged skin (Uitto et al, 1989). In a study of collagen production in chronologically aged skin, the content of type I collagen, the major collagen in the skin and a marker of collagen synthesis, is deceased by 68% in old skin versus young skin, and cultured young fibroblasts synthesize more type I collagen than old cells (Varani et al, 2006). The main reason for decreased production of collagen was found to be due to decreased synthesis of mRNA for type I collagen, and there was a three-fold reduction in the steady-state level of type I collagen mRNA in senescent fibroblasts (Furth, 1991). More fundamental mechanisms for age-related collagen synthesis involve the transforming growth factor-β (TGF-β)-induced signaling pathway of collagen synthesis (Figure 1). The ability of TGF-β to stimulate the synthesis of extracelluar matrix components in cultured fibroblasts is well-documented (Ignotz and Massague, 1986; Raghow et al, 1987; Bitzer et al, 2000). TGF-β-induced Smad2 phosphorylation is an initial molecular event mediated by TGF-β receptor I kinase. Phosphorylated Smad2 dissociates from the receptor and then forms a complex with Smad4. The Smad complex translocates to the nucleus where it activates the transcription of target genes including COL1A2, a gene for type I procollagen. In the early step of this process, Smad2 phosphorylation may also occur as the result of activation of a kinase located downstream of MEK-1 and upstream of the MAPK/ERK pathway (Brown et al, 1999). Other intermediate factors may be involved in this signaling pathway; one protein that induces the gene expression of extracellular matrix protein is the connective tissue growth factor (CTGF), a cystein-rich peptide (Grotendorst, 1997). CTGF is secreted by fibroblast cells after activation by TGF-β and acts as a downstream mediator of TGF-β-induced collagen synthesis (Duncan et al, 1999). It has been recently reported that CTGF is potently induced by TGF-β and stimulates type I procollagen expression through COL1A2 promotor activation (Holmes et al, 2001; Gore-Hyer et al, 2002). Accordingly, the TGF-β/Smad/CTGF/procollagen axis is the main signaling pathway for collagen synthesis in dermal fibroblasts and it is also found that TGF-β, CTGF, and type I procollagen genes are all downregulated in aged human skin in vivo (Quan et al, 2010). In this study, expression levels of TGF-β, CTGF, and type I procollagen genes were reduced to 70, 50, and 75%, respectively, in aged dermis compared to young dermis. Therefore, reduced synthesis of collagen in aged skin can be clearly explained by downregulation of signaling proteins in TGF-β-mediated collagen synthesis. The loss of collagen synthesis is recognized as a characteristic of chronologically aged skin by the cellular senescence of fibroblasts. However, there are some reports providing evidence that collagen synthesis is decreased by UV irradiation, the major factor for extrinsic skin aging (Fisher et al, 2000; Quan et al, 2004). The mechanism of reduced collagen production by UV irradiation is also related to the TGF-β/Smad pathway (Figure 1). In human skin, UV irradiation impairs the TGF-β/Smad pathway by downregulating TGF-β type II receptor, and leads to reduced TGF-β responsiveness and repression of TGF-β target genes including type I procollagen (Quan et al, 2001; Quan et al, 2006). UV irradiation can also reduce collagen production in dermal fibroblasts by a different mechanism associated with mechanical tension. According to previous studies, fibroblasts in healthy cells have normal mechanical tension by attaching to intact collagen fibrils and containing abundant actin in their cytoplasm (Grinnell, 2000, 2003). In contrast, cells in photoaged skin are in a mechanically relaxed state from contacting fragmented or amorphous collagen and having lower amounts of actin. With reduced mechanical tension, signaling through MAPK or TGF-β is not effectively transduced to the nucleus and subsequent transcription of collagens genes is inhibited (Grinnell et al, 1999; Varani et al, 2004).
Increased degradation of the collagenous matrix
Another key condition of skin aging associated with atrophy of the dermal connective tissue is the destruction of extracellular matrix (ECM) components, in particular collagen fibers. Whereas both intrinsic and extrinsic factors are related with this destructive phenomenon, photoaging by UV irradiation has a much more substantial effect than other factors and the underlying mechanisms have been extensively investigated in many studies. In brief, the underlying molecular mechanism of photoaging includes the following processes. The first step is the generation of reactive oxygen species (ROS) by UV irradiation (Black, 1987). ROS, including superoxide anions, hydrogen peroxide, hydroxyl radicals, and singlet oxygen (Masaki, 2010) can activate growth factor and cytokine receptors in fibroblasts (Sardy, 2009). Activated receptors stimulate members of very complex MAPK signaling pathways such as AKT, JNK, ERK, and p38. Next, the activation of NF-κB, c-Jun, and c-Fos successively follows in the nucleus and the latter two proteins combine together to form AP- 1, the transcription factor which stimulates the transcription of matrix metalloproteinase (MMP) genes (Pimienta and Pascual, 2007; Yaar and Gilchrest, 2007). MMPs are ubiquitous endopeptidases responsible for the turnover and degradation of ECM, the main target molecules being ECM proteins including all types collagens, elastins, and proteoglycans (Nagase et al, 2006). AP-1 also inhibits procollagen gene expression by blocking TGF-β type II receptor/Smad signaling (Quan et al, 2004). Consequentially, UV irradiation gives rise to dermal damage from collagen degradation by increased expression of MMPs and inhibition of procollagen synthesis (Figure 1). Therefore, the accumulation of dermal damage by repeated sun exposure may result in characteristic wrinkling of photoaged skin. Intrinsic or chronological aging can also contribute to the collagen damage even though its influence is less potent than that of photoaging. The main causative agent in this situation is also ROS that are produced as a consequence of oxidative metabolism (Sohal and Weindruch, 1996). ROS are inevitably created during the process of mitochondrial oxidative energy generation and are considered to be the cause of intrinsic aging by the destruction of important cellular constituents including proteins, lipids, and DNA (Muscari et al, 1996; Sohal and Weindruch, 1996; Masaki, 2010). ROS also accumulate due to the reduced antioxidant capacity of aged cells as a result of cellular senescence (Kohen and Gati, 2000) and act as a common molecular mediator for both over-expression of MMPs and reduction of type I procollagen seen in photoaging-induced dermal damage.
Mouse models of skin aging
Various phenomena and mechanisms of skin aging have been verified by in vivo experimental data from mouse models. Mice are useful models for studying cutaneous aging as they are genetically similar to humans, affordable for experiments, and easy to manage. They are particularly favorable for investigating intrinsic skin aging because of problems associated with reliable sampling and effects of the environment in human studies (Bhattacharyya and Thomas, 2004). Therefore, the changes of connective tissue by intrinsic or chronological aging have been confirmed in many mouse models (Fornieri et al, 1989). In one study of changes in collagen synthesis and degradation in male Lewis rat models between the ages of 1 and 24 months, collagen synthesis rates in skin at 24 months of age had decreased by at least 10-fold compared to rates in 1-month old animals and the proportion of newly synthesized collagen degraded in the skin was increased from 6.4% at 1 month of age to 56% at 15 months of age (Mays et al, 1991). In another report, total collagen synthesis in mouse skin expressed as the amount of hydroxyproline according to the wet weight was decreased by about 30% between 2 and 22 months of age (Boyer et al, 1991). Different results are available from histomorphological observations in the CBA mouse model in which a notable attrition of epidermal thickness and the number of epidermal cells could be correlated with age, but no conspicuous changes were noted in the depth of the dermis and the percentage area of collagen in aged mice (Bhattacharyya and Thomas, 2004). In another study with rats, dermal thickness remained constant from ages 2 to 22 months in Fisher rats while the dermis width increased until 1 year of age and thereafter remained constant in Wistar rats (Sundberg and King, 2001). These morphological observations rather than biochemical data do not always corroborate with in vivo mouse model experiments. That is because there are inherent differences in genetic constitution among different strains of colony-raised rats and mice, and technical factors that influence morphometric microscopic evaluation (Bhattacharyya and Thomas, 2004).
Mouse models can be also effectively used for elucidating the roles of ECM components in the dermis because of the possibility of transgenic modification for examining a target gene. As an example, a study of collagen-associated protein dermatopontin knockout mice represented the involvement of dermapontin in the formation of collagen fibrils (Takeda et al, 2002). In this study, the dermatopontin-null mice showed that the skin collagen content was 40% lower and the initial elastic modulus was 57% lower compared to wild-type mice. Collagen fibrils also showed great variation in diameter along with irregular contours. These data indicate that dermatopontin plays a critical role in skin elasticity and collagen fibrillogenesis in vivo. Phenotypic abnormalities have also been observed in various transgenic models for other ECM proteins such as decorin (Danielson et al, 1997), fibromodulin-(Svensson et al, 1999) and thrombospondin-2 (Kyriakides et al, 1998) null mutant mice that show skin laxity and fragility, and collagen fibril abnormalities. These transgenic mouse models are helpful to understand not only the roles of these proteins in dermal connective tissue but also the skin aging process because expression of these proteins is down-regulated with age.
Mouse models have been extensively employed for investigating photoaging as well. Studies of UV radiation-induced connective tissue damage in mouse models were first attempted in the 1960s. In the early stage, normal mice were used but hairless mice were adopted in the 1980s because normal mice with fur were not an appropriate model for human responses to UV irradiation. Currently, the most commonly used hairless mouse for studying photoaging is the albino Skh-hairless-1 (Kligman, 1996). The common feature of various alterations found in the dermal connective tissue following UV irradiation is elastosis, an accumulation of amorphous elastin material replacing the normal dermis (Baumann, 2007). Elastotic changes mainly appear in photoaged human and mouse skin, and are considered to be an important influence on the mechanical property of skin as well as the putative reason for the development of wrinkles (Carneiro et al, 2007). Other dermal changes, such as type I/III collagen and total collagen contents, induced by UV irradiation have been reported differently in several previous studies. In the report by Kligman et al, there were increases in total collagen in UV-irradiated hairless mice (Kligman et al, 1989). Additionally, in an experiment with photoaged C3H/HeN, C3H/HeJ, and Balb/c mice, UVB irradiation led to an increase in collagen, elastin, and glycosaminoglycans in all three strains (Kochevar et al, 1994). However, in another study the intact ultrastructural appearance of dermal collagen fiber bundles (DCFB) was gradually lost with increasing UV dosage in UV-irradiated hairless mouse skin and changes of the percent area of wrinkles was significantly correlated with degenerative changes of DCFB (Nishimori et al, 2001). This result concurs with the histologic and quantitative study of human facial skin by Warren et al (1991) in which a group with higher sun exposure had more wrinkles, more severe elastosis, and less collagen than a group with less sun exposure. Because of the complexity of skin organization and the genetic diversity among various strains of rats and mice, different histomorphologic and biochemical studies of the aging process in skin may only give partial pictures of overall cutaneous senescence. Notwithstanding, mouse models have been adopted and developed effectively for studying the mechanisms of skin aging and testing the effects of pharmacological tools to retard this process.
Summary
The phenotypes of aged skin such as wrinkling, laxity, sagging, and fragility have become a target for anti-aging studies from a standpoint of the skin's role as a social interface as well as its physiological function. The aging process in skin is a more complex process influenced by intrinsic and extrinsic factors than that of any other body organs, and the effects of these two types of factors overlap for the most part. Dermal matrix alterations are also affected by the combined effects of these two aging processes. Studies of skin aging have been extensively performed to elucidate the underlying mechanisms and to develop anti-aging measures. For this, various mouse models have been used to establish the processes of skin aging through experimental modifications. Although mouse models do not always corroborate the biochemical data and produce consistent results, they are the utmost useful tools to examine the fundamental etiology of skin aging and anti-aging effects.
 XML Download
XML Download