

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The human respiratory tract is constantly exposed to harmful microbes and air pollutants. The immune system responds to these factors to protect the host, but an unbalanced inflammatory response can promote tissue damage and ultimately lead to acute and chronic respiratory diseases.1 Asthma is a disease of the conducting airways that involves various degrees of airflow obstruction and airway hyperresponsiveness (AHR).2 Inflammation in asthmatic lungs contributes to the pathophysiology of the disease. This occurs mostly through the release of inflammatory mediators and remodeling of the airways. Inflammation is a fundamental innate immune response to environmental factors, including air pollutants.3 Accumulating evidence indicates that the inflammasome plays a key role in the pathogenesis of acute and chronic respiratory diseases.1 The inflammasome also plays a role in chronic inflammation of the airways of patients with asthma and chronic obstructive pulmonary disease as well as in the initiation and progression of the inflammatory process in pulmonary fibrosis.4 The inflammasome is a multiprotein complex that regulates inflammation by activating specific proinflammatory cytokines, resulting in an effective host immune response.5 The innate immune system is the first line of host defense, in which the inflammasome is essential for maintaining a delicate balance between pro- and anti-inflammatory signals to generate an appropriate immune response without harming the host.5 Activation of the multiprotein complex inflammasome by danger signals plays diverse and sometimes conflicting suppressive or stimulatory roles in cancer development and progression, with marked context-dependency.3 The inflammasome is a major regulator of inflammation through its activation of pro-caspase-1, which cleaves pro-interleukin-1β (pro-IL-1β) into its mature form. IL-1β is a critical pro-inflammatory cytokine that dictates the severity of inflammation associated with a wide spectrum of inflammatory diseases. NAIP, CIITA, HET-E, TP-2 (NACHT), leucine-rich repeat (LRR) and pyrin domain-containing protein 3 (NLRP3) are key components of the inflammasome complex, and multiple signals and stimuli trigger formation of the NLRP3 inflammasome complex.6
Nanoparticles (NPs)7 are produced in many natural processes and human activities, such as those involving automobiles, industry, and charcoal burning, and they profoundly affect air quality worldwide.89 NPs are associated with asthma exacerbation and asthma immune responses.101112
To date, the role of the inflammasome in the pathogenesis of asthma-related NPs has received limited attention. In this study, we investigated the influence of NPs on the inflammasome, AHR, and inflammation in a mouse model of ovalbumin (OVA)-induced allergic lung disease.
MATERIALS AND METHODS
Experimental design
Eight BALB/c mice were exposed to saline (sham) or OVA or OVA plus titanium dioxide (TiO2) NPs (50 µg/m3) (Standard Reference Material [SRM] 1898; National Institute of Standards and Technology [NIST], Gaithersburg, MD, USA) (Fig. 1) with some modifications of previous animal models.1314 After sonication, the endotoxin concentration of the diesel exhaust particle (DEP) suspension was less than 0.064 ng/mL (0.32 EU/mL), based on a Limulus Amebocyte Lysate assay (QCL-1000; BioWhittaker, Walkersville, MD, USA).15 The endotoxin concentration of the NPs was less than 0.064 ng/mL (0.32 EU/mL), based on a Limulus Amebocyte Lysate assay (QCL-1000; BioWhittaker).15 Analyses included measurement of IL-1β, IL-18, NLRP3, caspase-1, and lung protein levels as well as of bronchoalveolar lavage (BAL) fluid.
Animal exposure protocols
Female, 6-week-old BALB/c mice were sensitized by intraperitoneal (IP) injection on days 0 and 14 with 50 µg of grade V chicken egg OVA (Sigma-Aldrich, St. Louis, MO, USA) emulsified in 10 mg of hydroxyl aluminum plus 100 µL of Dulbecco's phosphate-buffered saline (D-PBS). On days 21 to 23, all mice received intranasal (IN) challenge with 150 µg of grade III OVA (Sigma-Aldrich) in 50 µL of D-PBS. Control mice were sensitized and challenged with saline. Mice in the TiO2 NP groups were administered 50 µg/m3 NPs by inhalation at 2 hours before OVA challenge daily for 3 days. On day 23, AHR was measured, BAL fluid was collected, and lung tissue was processed for protein and reactive oxygen species (ROS) measurement and Diff-Quick staining. This study was approved by the Soonchunhyang University's Institutional Animal Care and Use Committee (IRB No.; SCHBC-Animal-2014-013).
Determination of airway responsiveness to methacholine
Airway responsiveness was measured in unrestrained, conscious mice 1 day after the last challenge, as previously described.1617 Mice were placed in a barometric plethysmographic chamber (All Medicus Co., Anyang, Korea), and baseline readings were taken for 3 minutes and averaged. Increasing concentrations of aerosolized methacholine, from 2.5 to 100 mg/mL, were nebulized through an inlet of the main chamber for 3 minutes. Readings were taken for 3 minutes and averaged after each nebulization, and the enhanced pause (Penh) was determined. Penh, calculated as (expiratory time/relaxation time-1)×(peak expiratory flow/peak inspiratory flow) is a dimensionless value that represents a function of the proportion of maximal expiratory to maximal inspiratory box pressure signals and is a function of the timing of expiration. Penh was used as a measure of airway responsiveness to methacholine. Results are expressed as the percent increase in Penh following challenge with each concentration of methacholine, and baseline Penh (after saline challenge) is expressed as the actual value. Penh values were evaluated for 3 minutes after each nebulization and averaged.
BALF morphology analysis
BAL was performed 4 times using 1 mL of normal saline with gentle retrieval. Cells were counted using a hemocytometer, and differential cell counts were performed on slides prepared by cyto-centrifugation and Diff-Quik staining (Scientific Products, Gibbstown, NJ, USA).
Western blotting
Extracted lung tissue was homogenized in protein lysis solution containing 50 mM Tris-HCl (pH 7.4), 50 mM NaCl, 0.1% sodium dodecyl sulfate (SDS), 1% TritonX-100, 0.5 mM ethylenediaminetetracetic acid (EDTA), and 100 mM phenylmethylsulfonyl fluoride (PMSF) in distilled water, centrifuged at 14,000 rpm at 4℃ for 30 minutes, and the soluble material collected. Lung tissue plasma membrane protein was extracted using a MEM kit (Thermo, Rockford, IL, USA). Mouse lung proteins were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked in 5% bovine serum albumin (BSA) containing 0.1% Tween 20 in Tris-buffered saline (TBS) for 2 hours at room temperature. The membranes were then incubated with rabbit anti-NRLP3 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-IL-18 (1:500; Santa Cruz Biotechnology), rabbit anti-IL-1β (1:1,000; Santa Cruz Biotechnology), and mouse anti-caspase-1 (1:2,000; Adipogen, San Diego, CA, USA) overnight at 4℃. Membranes were next incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies. Detection was performed using the WEST-ZOL plus the Western Blot Detection System (iNtRon, Seongnam, Korea). Relative protein abundance was determined by quantitative densitometry; data were normalized to β-actin (Sigma-Aldrich), and plasma-membrane protein levels were normalized to that of Na/K ATPase.
Immunohistochemical analysis
Mouse lung sections were deparaffinized and rehydrated in an ethanol series. The sections were treated with 1.4% H2O2 in methanol for 30 minutes to block endogenous peroxidase; they were then non-specific binding blocked using 1.5% horse serum and incubated with mouse anti-caspase-1 (1:2,000; Adipogen). The following day, sections were incubated with an ABC kit (Vector Laboratories, Burlingame, CA, USA). Color reaction was developed using a liquid DAB+ substrate kit (Golden Bridge International Inc., Mukilteo, WA, USA). After immunohistochemical staining, the slides were counterstained with Herris's hematoxylin for 1 minute. Expression was quantified using the ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Enzyme-linked immunosorbent assay (ELISA) for 8-isoprostane and carbonyl products of ROS
We indirectly measured 8-isoprostane and carbonyl levels using ELISA kits (Cell Biolabs, San Diego, CA, USA). Extracted protein from lung tissue was treated with 10 N NaOH at 45℃ for 2 hours, and then 10 N HCl was added. The samples were centrifuged for 5 minutes at 12,000 rpm in a microcentrifuge, and the clarified supernatants were assayed. Protein was derivatized using dinitrophenylhydrazine solution and incubated at room temperature in the dark. After 1 hour of incubation at room temperature on an orbital shaker, anti-dinitrophenyl (DNP) antibody was added, followed by an HRP-conjugated secondary antibody. Finally, substrate and stop solution were added. Absorbance was read using a 450-nm filter and a microplate reader. Eight-isoprostane and carbonyl levels were assayed using an OxiSelect™ competitive ELISA (Mybiosource inc., San Diego, CA, USA). Eight-isoprostane and carbonyl cutoff values of 0.2 and 10 µg/mL, respectively, were used according to the manufacturer's recommendations.
RESULTS
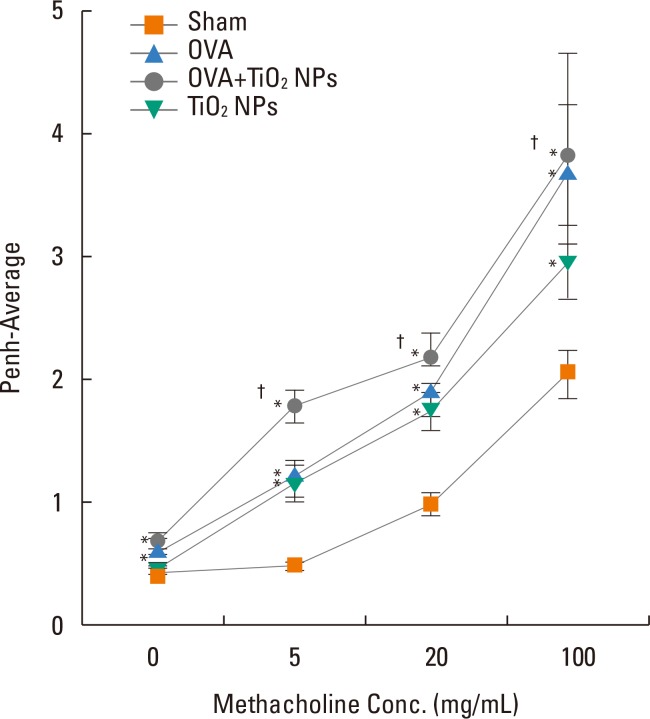
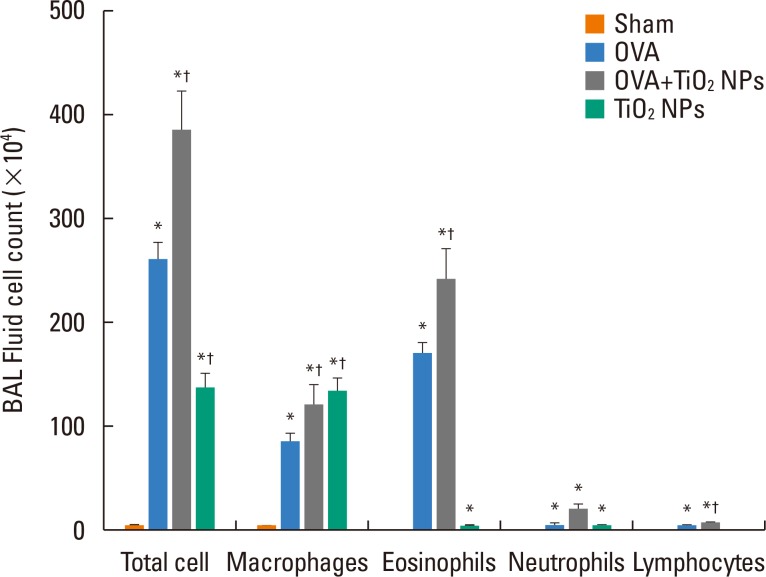
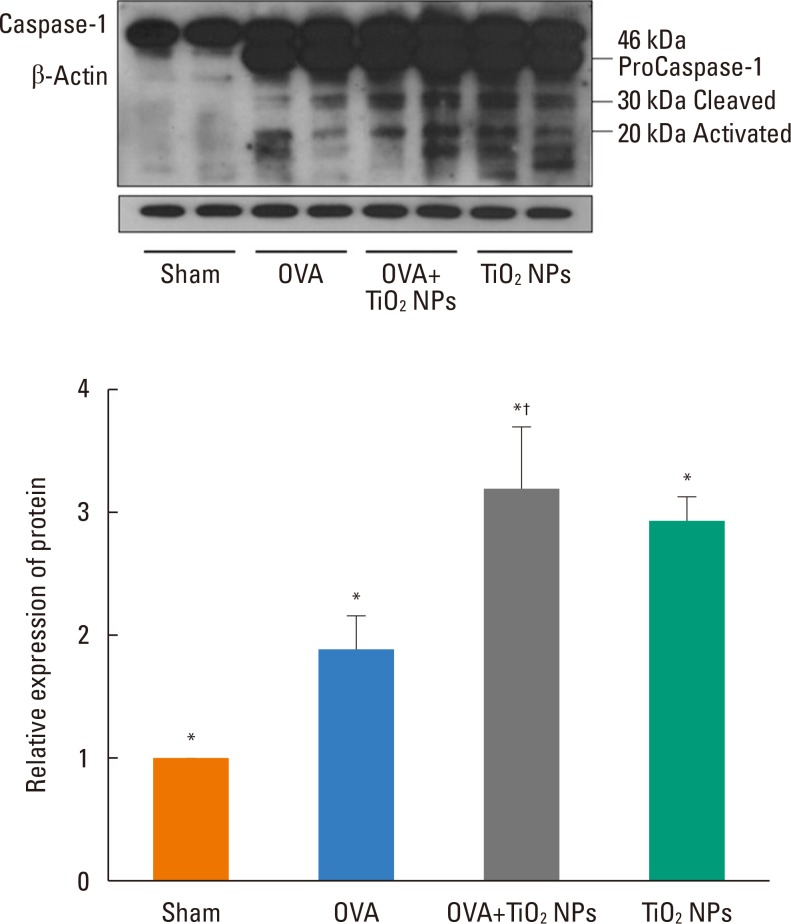
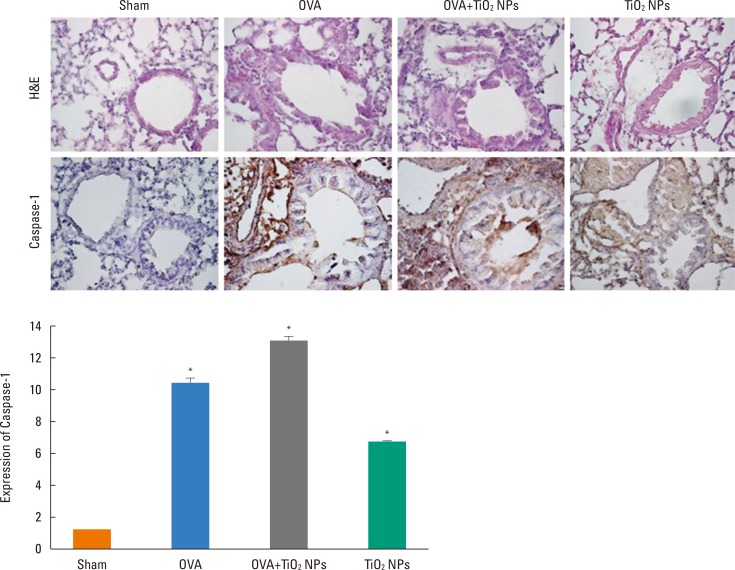
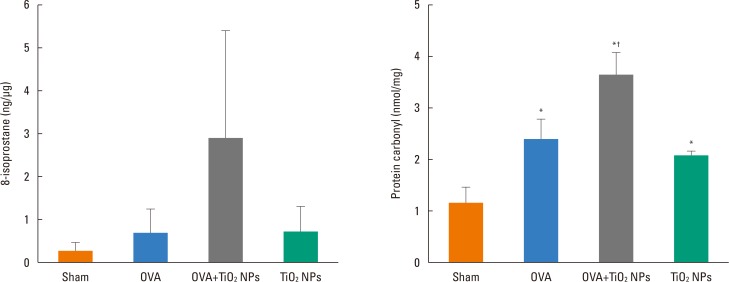
TiO2 NP-exposed mice exhibited a greater increase in Penh compared with saline-treated mice (P<0.05, Fig. 2). OVA-exposed mice exhibited a greater increase in Penh compared with saline-treated mice, and this was exacerbated by TiO2 NP treatment (P<0.05, Fig. 2). BAL fluid showed increased total and differential cell counts in the TiO2 NP-exposed group compared with the saline-treated group. Moreover, neutrophil, eosinophil, and lymphocyte counts were increased in the TiO2 NP-exposed group compared with the saline-treated group (P<0.05, Fig. 3). The OVA-exposed group exhibited greater increases in total cell and differential cell counts compared with the saline-treated group, and these increases were exacerbated by TiO2 NP treatment (P<0.05, Fig. 3). TiO2 NP treatment increased IL-1β, IL-18, and caspase-1 protein levels, but not NLRP3 protein levels in TiO2 NP-exposed mice (P<0.05, Fig. 4). The OVA-exposed group exhibited greater IL-1β, IL-18, and NLRP3 expression levels compared with the saline-treated group, and these were exacerbated by TiO2 NP treatment (P<0.05, Fig. 4). Activated caspase-1 expression in the lungs was increased in TiO2 NP-treated mice compared with saline-treated mice (Fig. 5). The OVA-exposed group showed greater activated caspase-1 expression compared with the saline-treated group, and this was augmented by TiO2 NP treatment (Fig. 5). Caspase-1 expression was increased in the lungs of TiO2 NP-treated mice compared with saline-treated mice (Fig. 6). The OVA-exposed group showed greater activated caspase-1 expression compared with the saline-treated group, and this was augmented by TiO2 NP treatment (Fig. 6). ROS levels in the lungs tended to be increased in TiO2 NP-exposed, OVA sensitized/challenged, and OVA-sensitized/challenged-plus-TiO2 NP-exposed mice (Fig. 7).
DISCUSSION
The increased prevalence of asthma suggests that environmentally derived cofactors, such as air pollutants, are important in the development and persistence of asthma.18 The respiratory tract is continually exposed to airborne matter, which translates into frequent opportunities for interactions with mucosa-associated immunity.18 The innate immune system is the first line of host defense, and the inflammasome is essential for maintaining the delicate balance between pro- and anti-inflammatory signals to generate an appropriate immune response without harming the host.19 The innate immune response encompasses cellular and non-cellular components. The airway epithelium and mucosal layer also mediate innate immune functions beyond serving as mechanical barriers. Innate immune cells include dendritic cells, innate lymphoid cells, and leukocytes, such as macrophages, neutrophils, and eosinophils.2021 Activation of the multiprotein inflammasome by NPs plays diverse and conflicting, suppressive and stimulatory roles in the development, progression, and exacerbation of respiratory disease.22 Patients with neutrophilic asthma have augmented expression of key inflammasome components and increased release of IL-1β.23 In human airways, a functional NLRP3 inflammasome has been identified in airway epithelial cells,24 and peripheral blood neutrophils express all components of the NLRP3 inflammasome, as well as IL-1β.25 The NLRP3 inflammasome is a major component of the innate immune system, but its mechanism of activation by a wide range of molecules remains largely unknown. TiO2 NPs induce pro-inflammatory cytokine and inflammasome activation through adenosine triphosphate (ATP), adenosine diphosphate (ADP), and adenosine.262728
In this study, TiO2 NPs increased IL-1β and IL-18 levels and led to airway inflammation and AHR, suggesting that NPs initiate an innate immune response in the airway in a mouse model of asthma.
NLRP3 is a key component of the inflammasome complex, formation of which is triggered by multiple signals and stimuli.2930 In this study, expression of the components of the NLRP3 inflammasome was significantly increased in OVA exposed mice compared with control mice, suggesting that the NLRP3 inflammasome is involved in the pathogenesis of asthma. NPs induced expression of inflammasome components, leading to the formation of an active inflammasome (caspase-1 activation) in the lung. This suggests that the NLRP3 inflammasome plays a role in the host response to TiO2 NPs and is a determinant of the clinical manifestations and exacerbations of asthma. This also explains the variable pathological presentations of asthma with respect to the inflammation process of TiO2 NPs. Further studies are needed to elucidate the effect of NPs in asthmatic patients.
Diesel and automobile exhausts are the primary sources of atmospheric nano- and micro-particles in urban areas.31 Most particles from vehicle exhaust are in the size range 20-130 nm for diesel engines and 20-60 nm for gasoline engines.323334353637 The NP concentration near freeways can be high for several hundred meters, showing that vehicular pollution is a major source of local contaminant particulate matter, including NPs. Humans and their activities generate considerable quantities of particulate matter indoors.38 In this study, NP exposure was found to increase AHR and airway inflammation, indicating that NPs can induce asthma. In particular, TiO2 NPs were found to increase AHR and airway inflammation in a mouse model of allergic asthma, indicating that NPs can exacerbate the respiratory symptoms of asthmatic patients.
ROS may serve as a ‘kindling’ or triggering factor to activate NLRP3 inflammasomes, as well as ‘bonfire’ or ‘effector’ molecules, resulting in pathological processes.5 ROS, which are produced by many known activators of NLRP3 inflammasomes, trigger NLRP3 inflammasome formation and activation in response to many exogenous stimuli as well as endogenously produced molecules or factors secreted by damaged cells, such as damage-associated molecular patterns (DAMPs).3940 In this study, ROS levels tended to be increased in the lungs of mice exposed to TiO2 NPs, OVA-sensitized/challenged, and OVA-sensitized/challenged-plus-TiO2 NP-exposed mice. This suggests that NPs can increase ROS levels and lead to increased expression of IL-1β, IL-18, and inflammasome pathway proteins, causing airway inflammation and hyperresponsiveness. In our study, the signaling pathway governing inflammasome formation in the lung was found to require a priming signal to activate NLRP3 and to induce caspase-1 activation and asthma (Fig. 8). Our data demonstrated that inflammasome activation occured in the lungs of a mouse model of allergic asthma following NP exposure, suggesting that targeting the inflammasome may assist in controlling NP-induced airway inflammation.



 XML Download
XML Download