

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Airway epithelial cells not only provide a physical barrier to potentially harmful insults, but they also play a crucial role in the first line of immunological defense.1 Human nasal epithelial cells are routinely used for clinical diagnostic purposes and have been shown to be good surrogate models in studies of nasal epithelium.12 Cultured airway epithelial cells are good research tools for studying airway epithelial physiology and have been instrumental in increasing our knowledge of the role of epithelial cells in airway diseases. To date, many kinds of epithelial cell culture methods, such as submerged, suspension, floating, and air-liquid interface (ALI), have been introduced and widely utilized for primary human airway epithelial cell culture. These cultures more closely resemble the physiology of in vivo conditions than immortalized cell lines.123456 Tissue for establishing primary airway epithelial cell cultures is usually obtained from transplant lung, or bronchial explants during surgeries and autopsies.7 Fresh tissues yield the greatest quantity of cells for culture, but obtaining tissues from such sources can be difficult on subjects, sometimes requiring surgery under general anesthesia for sampling. In particular, general anesthesia in diseased patients and children can be a high-risk procedure that may cause serious side effects or complications. Moreover, biopsied and de-epithelialized turbinate or polyp tissues are widely used for sampling nasal specimens from subjects. However, these tissues may not replicate the physiologic condition due to the preconditioning process required before the subculture.
The nasal brushing and culture technique has been introduced for cystic fibrosis patients instead of bronchial lavage and bronchial brushing.8 That technique is less invasive to patients, less expensive, and easier to perform than others. Also, intranasal brushing and direct cytology sampling are used to confirm the pathology of upper airway diseases and provide the ability to analyze the functional properties of airway cells. Although this technique does not appear to be widely used in the epithelial cell culture due to the possibility of contamination and low cellularity, we hypothesized that intranasal brushing and culture technique can be utilized for nasal epithelial cell study to determine the molecular phenomena that occur in diseased nasal epithelium.
In normal nasal epithelium, a delicate and tight balance of self-renewal and differentiation is regulated by key gene expression networks and molecular pathways.910 During inflammatory or environmental stress, the nasal epithelium frequently undergoes injury, followed by a rapid remodeling phase. These responses can include epithelial hyperplasia, goblet cell metaplasia, denudation, cilia loss, fibrosis or even basement membrane thickening.11 There is increasing evidence that allergic diseases, such as allergic rhinitis (AR), are associated with epithelial disorders and, furthermore, that primary abnormality of the airway epithelium may be central to causation and progression of AR.1213 Chronic inflammation and airway remodeling are the main characteristics of allergic diseases, and allergy-induced airway remodeling is characterized by goblet cell hyperplasia, reduced ciliated cells, mucus hypersecretion, defective repair and proliferation, increased basal cell number, and impaired barrier function.141516 AR is considered a Th2 cytokine-mediated nasal inflammation that is accompanied by accumulation of eosinophils and mast cells in the nasal mucosa and increased serum levels of antigen-specific IgE.17 The nasal epithelium, which is the first site of exposure to inhaled antigens, may play an essential role in the pathophysiology of AR, and it is thought that epithelial cell-derived cytokines, including thymic stromal lymphopoietin (TSLP), IL-25, and IL-33, are critical regulators of Th2 cytokine-mediated inflammation at nasal mucosal sites.18 It is well known that nasal epithelium characteristics and functions may provide an important insight for understanding the pathogenesis of AR, and the differentiated ALI culture model is the most appropriate platform with which to conduct in vitro research about AR.
In the present study, we compared the histologic and physiologic profiles of differentiated ALI cultures of nasal epithelial cells from healthy volunteers and those from AR patients. Moreover, we aimed to determine whether cultured nasal epithelial cells of AR also maintain allergy-induced disease characteristics and to assess feasibility for the study of epithelial functions in AR.
MATERIALS AND METHODS
Intranasal brushing and nasal cytology sampling
Subjects were recruited from outpatient clinics, and AR was diagnosed by allergic skin tests and the measurement of specific IgE levels using subjects' serum. All subjects were free of clinical signs of rhinosinusitis and respiratory infection, and had no history of other allergic diseases including asthma. Intranasal brushing was performed in 16 subjects under topical anesthesia, and nasal cytology specimens were obtained. Eight subjects, who tested positive in the allergic skin test and for the allergen-specific IgE, were diagnosed with AR and the other 8 subjects were classified as non-AR healthy subjects (Supplement Table). None of the patients had been on a regimen of intranasal medication, oral steroids, or antibiotic treatment for 3 months prior to the study. The Institutional Review Board of Yonsei University College of Medicine provided approval for this study (IRB# 4-2012-0136), and all adult subjects who participated in the study provided written informed consent.
A customized nasal brush (Vansco Korea, Daecheon) was inserted between the inferior turbinate and nasal septum, and the brush was gently spun 15-20 times at the middle portion of the inferior turbinate. The nasal cytology specimen and brush tips were immersed and transferred to transfer media (Dulbecco's Modified Eagle's Medium [DMEM]: HAM's F12K medium [F12]+1% Penicillin/Streptomycin) in 15-mL conical tubes. After vortexing of the conical tubes, tips were gently removed in a biological hood, and tubes were centrifuged at 1,200 rpm/min, 4℃ for 5 minutes. The supernatant was carefully aspirated, and the nasal cytology specimen was resuspended in 0.15% pronase solution (F12: 7 mL+pronase: 0.0105 g). Then, tubes were centrifuged at 1,200 rpm/min, 4℃ for 5 minutes, the supernatant was aspirated, and the specimen was resuspended in washing buffer (transfer buffer+10% fetal bovine serum). After pipetting or inverting several times, tubes were centrifuged at 1,200 rpm/min, 4℃ for 5 minutes. After aspirating the supernatant, the nasal cytology specimen was resuspended in subculture media and was plated to p-100 plates (passage 0).
Cell culture
Normal human nasal epithelial (NHNE) cells were cultured as previously described.46 Briefly, passage-2 NHNE cells (1×105 cells/culture) were seeded in 0.5 mL of culture medium on Transwell clear culture inserts (24.5-mm, with a 0.45-mm pore size; Costar Co., Cambridge, MA, USA). Cells were cultured in a 1:1 mixture of basal epithelial growth medium and DMEM containing previously described supplements.4 Cultures were grown while submerged for the first 9 days. The culture medium was changed on Day 1, and every other day thereafter. An ALI was created on Day 9 by removing the apical medium and feeding the cultures from the basal compartment only. The culture medium was changed daily after the initiation of the ALI. We added antibiotics such as 1% penicillin and streptomycin into the all media for subculture and culture stages and we also added antifungal agent, fungizone (1 mL/1,000 mL media) (Life technologies, Grand island, NY, USA) after filtering the media. All experiments described here used cultured nasal epithelial cells at 14 days after the creation of the ALI.
Real-Time PCR
Total RNA was isolated from nasal epithelial cells at 14 days after confluence, using TRIzol (Invitrogen, Waltham, MA, USA). The cDNA was synthesized from 3 mg of RNA with random hexamer primers, using Moloney Murine Leukemia Virus reverse transcriptase (PerkinElmer Life Sciences, Waltham, MA, USA and Roche Applied Science, Indianapolis, IN, USA). Commercial reagents (TaqMan Universal PCR Master Mix; PE Biosystems, Foster City, CA, USA) were selected, and conditions were set according to the manufacturer's protocols. The total reaction volume of 12 mL contained 2 mL of cDNA (reverse transcription mixture), oligonucleotide primers at a final concentration of 800 nM, and the TaqMan hybridization probe at 200 nM. The real-time PCR probe was labeled at the 5' end with carboxylfluorescein, and at the 3' end with the quencher carboxytetramethylrhodamine. Primers for human Foxj1 (Hs00230 964_mL), TEKT1 (Hs00364985_mL), TSLP (Hs00263639_mL), IL-25 (Hs03044841_mL), IL-33 (Hs00369211_mL), E-cadherin (Hs01023894_mL), and ZO-1 (Hx01551861_mL) were purchased from Applied Biosystems (Foster City, CA, USA). Real-time PCR was performed using the PE Biosystems ABI Prism 7700 Sequence Detection System. The thermocycler parameters included 50℃ for 2 minutes and 95℃ for 10 minutes, followed by 40 cycles of 95℃ for 15 seconds and 60℃ for 1 minute. Target mRNA levels were quantified using target-specific primer and probe sets for Foxj1, TEKTIN-1, TSLP, IL-25, IL-33, E-cadherin, ZO-1, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). All PCR assays were quantitative, and used plasmids containing the target gene sequences as standards. All probes were designed to span an intron, and did not react with genomic DNA. All real-time PCR data were normalized to the level of GAPDH (1×106 copies) to correct for variations between samples. All reactions were performed in triplicate, and the results were normalized against GAPDH as an endogenous control.
Electromicroscopy
Electron microscopic analysis was performed, as previously described, with some modifications.4 Specimens were fixed with 2% glutaraldehyde and 2% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4 at 4℃ for 2 hours, were washed, and were then incubated with 1% osmium tetroxide in 0.1 M PB at 25℃ for 2 hours. For scanning electron microscopy (SEM), specimens were dehydrated in graded baths to 100% ethanol, were critical-point-dried under liquid carbon dioxide, gold sputter-coated, and were visualized on a Hitachi S-800 microscope (Hitachi, Tokyo, Japan). For transmission electron microscopy (TEM), osmium-stained samples were further fixed in 7% uranyl acetate, were thin-sectioned (70 nm) in Polybed 812 (Polysciences, Warrington, PA), post-stained in uranyl acetate and lead citrate, and were visualized on a JEM1011 microscope (JEOL, Tokyo, Japan).
Histologic analysis and immunohistochemistry
De-waxed sections were stained with either hematoxylin and eosin (H&E), or a periodic acid-Schiff (PAS) kit, according to the manufacturer's instructions (Bioptica, Milan, Italy). Immunostaining was performed on formalin-fixed paraffin sections (4 µm) of cultured NHNE cells and inferior turbinate specimens. Formalin-fixed paraffin sections (4 µm) were de-waxed in xylene (Sigma Chemicals, St Louis, MO, USA), were rehydrated in successive ethanol baths, and were subjected to antigen retrieval by microwave in 0.01 mol/L sodium citrate buffer (pH 6.0). Endogenous peroxidase activity was quenched with 3% methanolic hydrogen peroxide for 10 minutes at room temperature. Nonspecific binding was blocked by incubation with 10% normal serum from VECTASTAIN Elite ABC Kit (Vector Laboratories, Burlingame, CA, USA) for 30 minutes at room temperature. Primary antibodies (1:200 dilution) were applied at 4℃ for 24 hours. Following washing in TBS, slides were incubated with peroxidase-conjugated goat anti-mouse/rabbit antibodies (1:200 dilution, Vector Laboratories) for 30 minutes at room temperature. Signal was amplified using the indirect immunoperoxidase technique using the DAKO Envision kit (Dako, Kingsgrove, Australia). Tissue sections were counterstained with Gill's hematoxylin (Sigma Chemicals), were dehydrated, and were mounted with DPX (ProSciTech, Thuringowa, Australia). Staining was visualized using an Olympus UTV0.63XC microscope with the DP Controller software (Hamburg, Germany). The experiment was performed using NHNE cells and inferior turbinate specimens from 4 healthy volunteers.
Measurement of transepithelial electric resistance (TEER)
The integrity of junctional proteins of the nasal epithelium was determined by measuring TEER in NHNE cells at 7, 14, 21, and 28 days after confluence. TEER is a widely used indicator of permeability in vitro, and was measured using the EVOM® resistance meter and Endohm® chamber (World Precision Instruments, Sarasota, USA). The values for cell-covered filters are expressed in standard units of ohms per square centimeter after subtracting the resistance of blank filters and are presented as mean+SD. Cells treated with medium alone served as a control.
RESULTS
Morphologic characterization of cultured NHNE cells using nasal brushes from healthy volunteers
Nasal cytology specimens were obtained from 8 healthy volunteers using intranasal brushing, and ALI culture was successfully established. NHNE cells were grown for 14 days in medium containing retinoic acid to induce mucociliary differentiation. Nasal epithelial cells were cultured on collagen-coated p100 plates during the subculture period and reached 80% confluence within 7 days. Light microscopic findings showed optimal cell growth and proliferation in the subculture (passages 0 and 1) (Fig. 1A). After subculture, passage 2 cells were seeded at a mean density of 1×105 cells/well on Transwell clear culture inserts, and ALI culture was created after 14 days confluence (Fig. 1A). Confluent NHNE cells were small, polygonal, and tightly joined; they had the typical cobblestone morphology of epithelial cells. We measured TEER at 7, 14, 21, and 28 days after confluence to confirm the formation of intact tight junctions, and normal-ranged TEER values were observed from 7 days after confluence (Fig. 1B).
Histologic findings, including H&E, PAS, and immunofluorescence staining for α-tubulin, as a marker of cilia, showed that ALI cultures at 14 days after confluence were 4 to 5 layers in thickness. The cells in the ALI cultures were well-differentiated, stratified, columnar epithelial cells and included secretory and ciliated cells (Fig. 1C). When viewed using a light microscope at a higher magnification (×40), it was possible to observe cilia beating 14 days after confluence.
SEM of apical cells in ALI culture at 14 days after confluence showed polygonal cells tightly attached to each other, and well-differentiated cilia and secreted mucus were observed (Fig. 2A, B). TEM also showed ciliated and secretory cells that were well differentiated, with intact tight junctions (Fig. 2C, D).
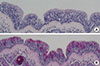
As the next step, we investigated the histologic findings of real human nasal mucosa to compare the morphologic characterization of nasal epithelium with cultured NHNE cells. Histologic examination using H&E and PAS staining was performed, and the results demonstrated that a multilayered nasal epithelium was observed including intensely stained ciliated cells and secretory cells (Fig. 3A, B).
Histologic findings revealed that ALI culture of well-differentiated NHNE cells was achieved using nasal cytology specimens and that cultured nasal epithelium had the histologic shape of real epithelium of human nasal mucosa. We estimated that intranasal brushing may be a useful method to perform NHNE cell culture and may be a very important tool for in vitro study about nasal epithelium.
Histologic examination of cultured nasal epithelial cells of AR patients
Intranasal brushings were performed in AR patients, and nasal epithelia cells were cultured with the same culture method and media as the ALI culture of NHNE cells.
We first measured the required period of subculture and confluence of ALI culture in ARNE cells. The mean period of subculture in NHNE cells was 5.9 days and that in ARNE cells was 6.1 days. The mean period from cell seeding of ALI culture to confluence was not significantly different between NHNE and ARNE cells culture (Fig. 4A, B). We found no overall differences in the rate at which the epithelial cells could proliferate and grow between normal subjects and AR patients, and it took about 28 days to obtain fully differentiated human nasal epithelial cells after intranasal brushing was performed.
We then carried out the histologic examination using ALI culture of ARNE cells at 14 days after confluence. Histologic examination, including H&E, PAS, and immunofluorescence staining for α-tubulin, showed significant dissimilar findings compared to the culture results of NHNE cells such as only 2 or 3 layers in thickness, more intense PAS staining, and fewer cilia at 14 days after confluence (Fig. 4C). We also quantified the number of secretory cells using PAS score and measured the number of ciliated cells through light microscopic findings. The results demonstrated that cultured ARNE cells had a higher PAS score (17.0+3.6 pixels/µM) than NHNE cells (6.0+1.8 pixels/µM), and a considerably smaller number of ciliated cells were observed in ARNE cells (ARNE cell culture: 11.6+3.3/HPF vs NHNE cell culture: 25.0+2.6/HPF). In addition, we measured the mRNA levels of Foxj1 and TEKTIN-1 in NHNE and ARNE cells at 14 days after confluence to assess the ciliogenesis and differentiation of ciliated cells. Interestingly, both mRNA levels of Foxj1 (ARNE cell culture: 19,332.5+3,464.1 vs NHNE cell culture: 5,119.5+1,017.2, Fig. 4F) and TEKTIN-1 (ARNE cell culture: 28,296.5+3,871.4 vs NHNE cell culture: 732.5+101.1, Fig. 4G) were significantly lower in cultured ARNE cells, although brushed samples from healthy volunteers and AR patients were cultured under the same culture conditions and media. These data suggest that there were significant differences in epithelial cell cultures from intranasal brushed specimen between NHNE and ARNE cell culture.
Electronic microscopic findings of cultured ARNE cells
SEM and TEM findings provided more detailed information about the histologic differences between cultured ARNE cells and NHNE cells. As expected, SEM findings showed that the number of ciliated cells was significantly attenuated in ALI culture of ARNE cells at 14 days after confluence (Fig. 5A), and abnormal cilia architectures were observed with more untidy and shorter cilia structures on the epithelial surface (Fig. 5B). There were also significantly different findings in TEM findings of ARNE cells, such as an increased number of secretory cells and loose tight junctions (Fig. 5C, D). In addition, both mRNA levels of E-cadherin (ARNE cell culture: 11,549.7+4,428.6 vs NHNE cell culture: 28,713.5+8,273.4, Fig. 5E) and ZO-1 (ARNE cell culture: 4,953.5+1,897.7 vs NHNE cell culture: 11,279.2+7,406.6, Fig. 5F) were significantly attenuated in cultured ARNE cells at 14 days after confluence under the same culture conditions. These findings suggest that loose tight junctions, a smaller number of ciliated cells, and less dense and shorter cilia were distinct characteristics of the nasal epithelium in AR patients and ALI culture of ARNE cells from nasal cytology specimens clearly showed different histologic features when compared to cultured NHNE cells.
The characterization of AR in ARNE cell culture
The allergic airway is characterized by the secretion of Th2 cytokines, which has been shown to be highly involved in the pathogenesis of allergic inflammation. In particular, TSLP, IL-25, and IL-33 can initiate allergic inflammation and are produced by several epithelial cell lines, keratinocyte, and respiratory epithelial cells. To evaluate if cultured ARNE cells, which had different histologic findings from NHNE cells, possessed allergic characteristics without any stimulation, we measured the gene expression of TSLP, IL-25, and IL-33. NHNE cells from healthy volunteers (n=8) and ARNE cells from AR patients (n=8) were cultured, and cell lysates were obtained at 14 days after confluence. The results of real-time PCR showed that the mean mRNA levels of TSLP from ARNE cells were considerably higher than that of NHNE cells, although there was not any allergic stimulation to either type of nasal epithelial cells (NHNE cell culture: 1,637.2+815.1 vs ARNE cell culture: 4,115.4+708.5, Fig. 6A). However, mRNA levels of IL-25 and IL-33 were not elevated in ALI culture of ARNE cells (Fig. 6B, C). These results demonstrate that cultured ARNE cells from AR patients may have higher levels of Th2-initiating epithelium-derived cytokines, such as TSLP, and ALI culture of AHNE cells, acquired using intranasal brushing, preserved the Th2-related immune response resulting in the appearance of AR-related histologic characteristics.
DISCUSSION
Culturing NHNE cells and understanding the method of primary epithelial cell culture is not only critical for elucidating epithelial physiologic function, but also helps unravel the pathophysiology of numerous related diseases. The limitations imposed by the use of nasal epithelial cells are: the limited amount of specimens available from one donor; the possibility of contamination by pathogens; and their accessibility, which requires anesthesia and intubation. To date, it remains unclear whether differentiated nasal ALI cultures can be achieved using nasal brushing and whether ALI cultures from nasal cytology specimens maintain the disease-specific characteristics after full differentiation, especially AR.
In the present study, we used intranasal brushing to collectnasal specimens for ALI culture of nasal epithelial cells under topical anesthesia. We found that culturing nasal epithelium using nasal cytology specimen produced a typical epithelial cell phenotype when examined under light and electron microscopes, including histologic or immunofluorescence staining. Intranasal brushing may be a useful source for airway epithelial cell culture compared to the more aggressive bronchial brushing and tissue sampling from intranasal turbinate mucosa or nasal polyp; there was no discomfort experienced by the volunteers during or after brushing. Primary human cell cultures are sometimes preferred over immortalized cell lines since they are physiologically more similar to in vivo tissues, but challenges are associated with growing primary cells including limitation of sample accessibility, difficulty in cell isolation, contamination and side effects in volunteers that undergo the procedure. We found that nasal epithelial cell culture using nasal-brushed specimens was relatively simple, safe, and successful. Therefore, intranasal brushing can substitute for in vitro NHNE cell culture using nasal mucosa or nasal polyp obtained invasively during an operation under anesthesia.
Our data demonstrated that fully differentiated nasal epithelium, including ciliated and secretory cells, could be obtained by ALI culture using nasal cytology specimens and showed the feasibility of intranasal brushing in experimental research of the nasal epithelium. The sample contamination is the well-known limitation of cell culture using intranasal cytology specimens. In the present study, we disinfect all brushes before obtaining nasal samples and transferred to sterilized transfer media and also add antibiotics and antifungal agents after filtering the media. We think that addition of antibiotics and antifungal agent to media would be necessary to prevent the contamination of cultured NHNE cells.
The majority of in vitro studies have been conducted during the subculture stage or the early stage of ALI culture; however, the state of differentiation, cell phenotype, and the expression of genes for mucus production may differ according to the culture duration or method. Therefore, to obtain more reliable results, it is critical to investigate the state of cell differentiation, phenotype, and expression of various genes during the late stages of the culture. In the present study, the fully differentiated appearance of the nasal epithelium was observed 14 days after confluence and such cultures not only mimic the morphologic features of the stratified columnar epithelium, including secretory cell differentiation, but also retain morphologic characteristics of cilia, which are similar to the histologic findings of real nasal epithelium in human nasal mucosa.
The nasal epithelium not only serves as a mechanical barrier to protect against environmental factors, microorganisms, allergens, and toxins, but also participates in both innate and adaptive immune responses.910 Indeed, the primary target of allergens is the nasal epithelium, the functions and integrity of which are profoundly altered by allergen-induced cytokines in AR. In addition, better understanding of the pathophysiological mechanisms involved in AR requires the analysis of nasal epithelium obtained from human samples and the culture of nasal epithelial cells provides an attractive alternative for AR research.19
In the present study, we performed intranasal brushing in healthy volunteers and AR patients under topical anesthesia and compared the histologic findings of the ALI cultures between normal and AR epithelia. We found that the speed of cellular proliferation and pace of the differentiation in ALI culture of ARNE cells were not significantly different from ALI cultures of NHNE during subculture or culture stages. Fully differentiated ARNE cells were observed within 14 days after confluence with a normal growing rate, and culturing ARNE cells took at least 24 days, including an approximately 7 day subculture period.
We obtained fully differentiated ALI culture of ARNE cells at 14 days after confluence. Histological examinations revealed that a smaller number of cilia on the apical surface of the nasal epithelium, loose tight junctions, and hyperplasia of secretory cells were characteristics of cultured ARNE cells. In particular, the attenuated cilia of ARNE cells was the most outstanding finding in the ALI culture of ARNE cells and we have further analyzed 2 important genes, Foxj1 and TEKTIN-1, which are involved in ciliogenesis to better understand the changes observed in cultured epithelium of AR patients. Both genes are well known regulators of differentiation of cilia and the formation of motile cilia, and participate in multiple steps during cilia formation including centrosome multiplication, docking, and cilia elongation.20 High expression of Foxj1 and TEKTIN-1 can not only induce longer cilia, but also increase the number of cilia per cell. Therefore, down-regulation of both genes can result in the loss of cilia in the human airway. According to our results, the mRNA levels of Foxj1 and TEKTIN-1 were significantly attenuated in ALI culture of ARNE cells, and this was correlated with the smaller number of cilia in cultured nasal epithelial cells from AR patients. On the basis of our findings, we estimated that the progression of ciliogenesis might be abnormally down-regulated, and differentiation of secretory cells occurred more excessively in ALI culture of ARNE. Therefore, these histologically distinctive characteristics of ARNE cells contribute to dysfunction in mucociliary clearance and mucus hypersecretion in the nasal epithelium of AR patients.
TSLP, IL-25, and IL-33, which are produced by epithelial cells and other cell types, exert profound downstream effects on various immune cells.21 A growing body of literature suggests critical roles for epithelium-derived cytokines TSLP, IL-25, and IL-33 in the regulation of Th2-type immunity and allergic responses.22 Several recent advances in the biology of these cytokines point to shared functions as well as distinct characteristics, which cause allergic immune diseases, including allergic rhinitis. The pathophysiology of human TSLP, IL-25, and IL-33 involves innate allergic immune response with direct influence of those cytokines on mast cells or immature dendritic cells. TSLP-, IL-25-, and IL-33-activated mast cells or dendritic cells initiate adaptive allergic immune responses by triggering differentiation of naïve T cells into inflammatory Th2 cells that produce several traditional Th2 cytokines and large amounts of type I hypersensitivity-related cytokines.2122 Importantly, it has been reported that TSLP, IL-25, and IL-33 mRNA levels are increased in bronchial epithelial cells from severe asthmatic patients; the cytokines likely play important roles in the initiation, regulation and persistence of allergic asthma after secretion from bronchial epithelial cells.23
We presumed that these cytokines might be secreted in the nasal epithelial cells by biologic factors that are related to AR. However, to our knowledge, no specific information is available on the generation of TSLP, IL-25, and IL-33 in cultured nasal epithelial cells from AR subjects. In the present study, we found that the TSLP mRNA level was significantly higher in the ALI culture of ARNE cells compared to cultured NHNE cells, although the cells were not stimulated by any allergic cytokines after full differentiation. Unstimulated cultured ARNE cells maintained the distinguishing physiologic responses of secreted epithelium-derived allergic cytokines with TSLP, a key cytokine in the pathogenesis of AR, after ALI culture using nasal cytology specimens from AR patients. A plausible explanation for this may be that ALI culture of ARNE cells maintains the basic features of AR. Based on these findings, cultured ARNE cells from nasal cytology specimens of AR patients had significant differences from cultured nasal epithelial cells from healthy volunteers, and unique histologic or physiologic characteristics with AR were preserved. Therefore, it is feasible to study nasal epithelial function in AR and it will be necessary to investigate the unique mechanisms of AR in nasal epithelium. Interestingly, we did not observe any significant differences in IL-25 and IL-33 mRNA levels between ARNE and NHNE cells. The cellular mechanisms of IL-25 and IL-33 and regulation of both cytokines activities are not totally understood in the present study, but we presumed that TSLP gene expression in the epithelial compartments would be constitutively higher and be more dominant than IL-25 and IL-33 in nasal epithelium of patients who are diagnosed with AR. Although the tissue microenvironment likely plays a role, transcription of IL-25 and IL-33 and secretion from epithelial cells would be induced after allergen stimulation. It has been reported that IL-25 and IL-33 appear functionally similar, and the release of both cytokines may be dependent upon allergen exposure.24 We propose that the mRNA levels of IL-25 and IL-33 were not relatively higher in cultured nasal epithelium of AR patients before allergen stimulation, unlike TSLP, but could be released upon allergen stimulation in the cultured nasal epithelial cells. TSLP, IL-25 and IL-33, which are produced by epithelial cells, have a shared biological activity to induce Th2 cytokines, suggesting involvement in the development of allergic diseases, but play different pathogenic roles in nasal epithelium depending on allergen exposures.
In conclusion, the present study shows that, by means of minimally invasive nasal brushing, it is possible to harvest nasal cytology specimens in sufficient amounts for ALI culture of nasal epithelial cells in AR patients. ALI culture of ARNE cells showed distinguishing histologic morphologies and physiologic features compared to NHNE cells, and cultured ARNE cells preserved the pathologic conditions occurring in the nasal epithelium of AR patients. Such findings will expedite the clinical application of in vitro studies to predict the molecular mechanisms of AR and the potential role of the epithelium in allergic diseases.





 XML Download
XML Download