

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Lipid mediators participate in intercellular interactions as a paracrine signaling system which has evolved over a period of 600 million years in primitive metazoans: sponges and corals. In humans, these mediators contribute to any inflammation, and there is a pertinent question of how is this specific for asthmatic inflammation. Since the first description of the slow reaction smooth muscle-stimulating substance (SRS-A) by Feldberg and Kelloway 75 years ago, lipid mediators have been investigated in allergic reactions and bronchospasm. Advancements in clinical biochemistry made it possible to identify chemical structure, biosynthetic pathways, and cellular receptors of lipid mediators, and there were some novel ones identified during the last decade. These biological compounds became not only recognizable but also measurable in clinical samples due to development of affordable chromatography-mass spectrometry equipment or sensitive immunoassays. Some recent advances in understanding of the signaling by eicosanoid mediators during asthmatic inflammation are presented. Special attention is paid to aspirin-exacerbated respiratory disease (AERD), a phenotype of asthma manifested by the most profound changes in eicosanoids. Novel drugs inhibiting signaling or metabolism of eicosanoids are currently under development. It will require population studies to prove their efficacy in inflammation of the airways because contribution of eicosanoids to asthma is highly variable among asthmatics.
BIOSYNTHETIC PATHWAYS OF LIPID MEDIATORS
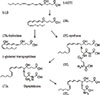
The most prevalent lipid mediators belong to the family of eicosanoids. This name describes 20 carbon backbones of abundant polyunsaturated fatty acids in the organism (Fig. 1). Among them, arachidonic acid, an omega-6 fatty acid with 4 polyunsaturated bonds, is a precursor of the most biologically potent lipid mediators. Biosynthetic pathway of eicosanoids involves either non-enzymatic or stereospecific enzyme-catalyzed oxidative reactions. Non-enzymatic oxidation affects polyunsaturated fatty acids at their storage site, which are esterified to phospholipids constituting the cell membrane. This oxidation may happen during any biochemical reaction producing free oxygen radicals in the organism. Particularly, it follows the oxidative burst of activated granulocytes. Produced oxylipins are named isoprostanes because their common signature is a stereochemical conformation different from mediators produced in enzymatic reactions. Isoprostanes are released from the cell membranes by the activity of phospholipases before they can exert any biological function. It is now widely accepted that isoprostanes activate the thromoboxane receptor and that their biological activity is similar to that of thromboxane A2.1 Specific enzymes, which oxidize free polyunsaturated fatty acids released by phospholipases, regulate rate-limiting production of eicosanoids (Fig. 1). These enzymes are known as lipoxygenases or cyclooxygenases. Different lipoxygenases preferentially oxidize polyunsaturated fatty acids at their certain carbons, as counted from the carboxyl group carbon, and some products named R or S may differ by stereochemistry. There are several human enzymes capable of oxidizing polyunsaturated fatty acids (Fig. 2). During inflammation, cellular 5-, 15, or 12-lipoxygenases are activated, and some studies have associated their genetic variants with particular phenotypes of inflammation.2 Another site-specific oxidation, at terminal ω-carbon number 20 is catalyzed by monooxygenases belonging to the cytochrome P450 family (CYP4A11, CYP4A22, CYP4F2, and CYP4F3) and contributes to the metabolic degradation pathway of eicosanoids.3 This takes place in the microsomal fraction of the liver or in the kidney and is accompanied by β-oxidation, shortening the carbon backbone of oxylipins. A direct product of lipoxygenation of arachidonic acid is a hydroperoxide of eicosatetraenoic acid (HpETE), but this intermediate is quickly reduced to an ordinary hydroxyl group of hydroxy-eicosatetraenoic acid (HETE). A more complex metabolism can occur downstream lipoxygenation pathways, involving production of glutathione conjugates (Fig. 3). Activity of 5-lipoxygenase (5-LO) results in production of 5-HPETE. This intermediate is oxidized again by the same enzyme to form leukotriene A4 (LTA4), a parent compound of the leukotriene family. Depending on the cell type and specific expression of enzymes, LTA4 is either hydrolyzed to a potent neutrophil chemoattractant leukotriene B4 (LTB4) or conjugated with glutathione to form leukotriene C4 (LTC4). LTC4 is equipotent with leukotriene D4 (LTD4) produced by γ-glutamyl transpeptidase. However, the most stable and the least potent is leukotriene E4 (LTE4) produced by dipeptidase cleavage of LTC4. Altogether these lipid mediators are named cysteinyl leukotrienes (CysLTs) and correspond to SRS-A formed in the allergic lung. Since LTA4 is easily diffusible across biological membranes, leukotrienes can be produced as transcellular mediators of inflammation. In the bloodstream, the most common transcellular metabolism occurs between activated granulocytes producing LTA4 and granulocyte adherent platelets expressing LTC4 synthase. Platelets do not have 5-LO activity, whereas neutrophils do not have LTC4 synthase; thus, biosynthesis of LTC4 is complemented only in neutrophil-platelet aggregates. An analogous family of vasoactive mediators produced by 15-LO and conjugated to glutathione is named eoxins,4 whereas cysteinyl-hepoxilins are produced from the 12-LO pathway.5
Prostanoids represent a subset of eicosanoid mediators produced by oxidation of polyunsaturated fatty acids in a cyclooxygenase pathway. Their chemical structure is distinguished by a common a 5 carbon cyclopentane ring, which is formed during the 2-step enzymatic activity of cyclooxygenases. There are 2 genes encoding cyclooxygenases (prostaglandin G synthases) in humans: PTGS1 and PTGS2. These enzymes are named cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2). Another protein encoded by the same PTGS1 gene was named PTGS-1b or COX-3. It has initially been described as translated from the splicing variant of PTGS1 transcript and particularly sensitive to acetaminophen inhibition with a limited tissue distribution. However, the latest report questioned the presence of enzymatically active COX-3 in humans and showed maturation of the PTGS-1b transcript to a normal COX-1.6 For simplicity, it is assumed that COX-1 expression is mostly constitutive, whereas COX-2 is an inducible enzyme. Both enzymes produce the same unstable intermediates: PGG2/H2, which are metabolized to the prostaglandins PGE2, and PGD2, and PGF2 by specific synthases. In the vascular system, PGH2 is also metabolized to thromboxane A2(TXA2) by platelets and to prostacyclin (PGI2) by endothelial cells.
Another group of eicosanoid mediators can be produced in the inflammatory milieu by metabolic steps dependent on intercellular cooperation. These are named pro-resolving eicosanoids because they terminate inflammatory reactions by activation of macrophage phagocytosis, without usual release of pro-inflammatory cytokines.7 It was recently discovered, that pro-resolving eicosanoids can also be conjugated to glutathione at the site of inflammation and retain a potent biological activity.89 Lipoxin (LXA4) was described as the first pro-resolving eicosanoid. Arachidonic acid is metabolized to lipoxins by a concerted action of lipoxygenases (5-LO + 12-LO for LXA4 and 5-LO + 15-LO for LXB4). Other polyunsaturated fatty acids, e.g., ω-3 docosahexaenoic acid, are similarly metabolized to protectins, resolvins, or maresins. It is interesting that following COX-1 inhibition by NSAIDs, the lipoxygenase activity of this enzyme is retained; however, stereoselectivity of the oxidation is altered. A series of aspirin-triggered 15-R lipoxins or their ω-3-derived analogs also has a potent anti-inflammatory activity.10
BIOLOGICAL ACTIVITY OF EICOSANOIDS
The biological activity of eicosanoids requires expression of their specific receptors. There are 2 receptors for CysLTs (CysLTR1 and CysLTR2), and 2 receptors for LTB4 (BLTR1 and BLTR2). Each prostaglandin has at least 1 specific receptor; however, there are 4 different PGE2 receptors (EP1-4) and 2 PGD2 receptors (DP1 and DP2).11 All these cellular proteins belong to a family of 7-transmembrane domain receptors, coupled with G proteins (G protein-coupled receptors [GPCR], Table). Pro-resolutionary eicosanoids also have their own receptors, e.g., FPR2/ALX. This explains a variety of biological effects caused by eicosanoids, which may either stimulate or inhibit cyclic AMP production by coupling with Gαs or Gαi subunits of the receptor, or release intracellular calcium by Gαo also leading to activation of protein kinases. A complex mechanism of signaling by eicosanoids, well studied on the cellular level, involves not only regulated biosynthesis of these mediators but also changes in their receptor densities.12 This may explain their highly variable biological effects in the organism, not always predictable using in vitro experiments.
EICOSANOIDS IN THE AIRWAYS
The lung has its own compartments for eicosanoid biosynthesis. Respiratory epithelium produces and releases eicosanoids in a polarized pattern. Prostaglandins are transported passively to the luminal surface, whereas HETEs are released to the basolateral one.13 Respiratory disorders are accompanied by disruption of the epithelial tight junctions and increased paracellular flux, or ultimately by increased shedding of epithelial cells accompanied by proliferation of epithelial progenitors.14 This might be reflected by changes in the local concentration of eicosanoids. During proliferation of epithelial cells, COX-2 activity is increased; however, it becomes down-regulated in the fully differentiated mucociliary epithelium. Under Th2-type conditions mimicked by interleukin (IL)-4 and IL-13, a potent induction of 15-HETE activity is observed, whereas 5-LO is present in differentiated cells, but not induction.13 These Th2-type cytokines also suppress PGE2 production; however, expression of COX-1 do not change and only COX-2 transcription decrease. Thus, respiratory epithelium has both cyclooxygenases activities. However, eicosanoids are also produced by other structural cells of the respiratory tract. Bronchial fibroblasts have activity of COX-1 and capability to induce COX-2.15 Airway smooth muscle cells also express prostanoid synthases and actively contribute to epithelial regulation producing PGE2.16 Inflammatory cells, infiltrating the lung, are an important source of eicosanoids, all expressing active metabolic pathways for eicosanoid biosynthesis.
CLINICAL STUDIES ON EICOSANOIDS IN ASTHMA
In clinical asthma studies, the most relevant are biological samples collected directly from the asthmatic airways. These include bronchoalveolar lavage fluid (BALF), induced sputum (IS), and exhaled breath condensate (EBC). Each material sampled has inherent advantages and drawbacks. BALF has to be collected during an invasive procedure of bronchofibroscopy, and there is no widely accepted method to compensate for a variable recovery of the instilled saline. This may lead to some dilution errors, usually not exceeding an order of magnitude in the measured concentration of mediators. IS collection procedure is time-consuming and requires a good cooperation of the patient, but is well tolerated. Induction of sputum is facilitated by inhalation of hypertonic saline solution, and is followed by immediate laboratory preprocessing during which expectorated mucus plugs are manually separated from saliva. Next, the sputum sample is weighted and solubilized using a constant proportion of a reducing agent solution (dithiothreitiol – [DTT]). Measurements of IS lipid mediators concentrations are more reproducible, but may be biased if the procedure is repeated within a short period of time. EBC is collected in a cooling trap as a condensed water vapor. It contains a small admixture of aerosol from the epithelial lining fluid. This aerosol is produced in the smaller diffusive airways by their collapse during inhalation and subsequent inflation causing detachment of the opposite airway surfaces during exhalation, following physiological changes in intrabronchial pressure. We learned that EBC can credibly reflect a baseline production of lipid mediators in the airways. However, the mechanism by which EBC carries admixture of the epithelial lining fluid aerosol is dependent on the pattern of ventilation. A decrease in the airflow during bronchoconstriction can change concentration of mediators.17 For example, a rise in eicosanoids is poorly reflected during a positive challenge procedure in aspirin-hypersensitive subjects. The upper respiratory tract, including nasal and paranasal cavities, has a histological structure of epithelium similar to the bronchi. Nasal lavage can be used as a source of surrogate material to study lipid mediators in asthmatics. It has the advantage that a time series of samples can be analyzed. It has been shown in AERD that CysLTs release reaches their maximal values in the nasal lavage 30 minutes after lysine-aspirin instillation, in parallel with the rise in eosinophil cationic protein (ECP). Symptoms of sneezing, nasal congestion and rhinorrhea appear much faster as the total nasal symptom score is already elevated after 10 minutes.18 In another study, increases in tryptase and PGD2 are also observed, whereas PGE2 decreasesas ECP increases.19
There are many problems in the assessment of the effect of eicosanoids on the systemic level. A simple venous blood sampling procedure can activate platelets and bias measurements of lipid mediators. If done properly, it requires collection using tubes with internal deuterated standards and anticoagulants immediately cooled on ice, followed by separation of plasma. Thus, it is commonly preferred to quantify eicosanoid metabolites in urine. However, prostaglandins, isoprostanes, and proresolutionary eicosanoids all undergo extensive inactivation before excretion with urine.20 The urinary products of their breakdown are β-oxidation metabolites. The most abundant urinary prostanoid metabolites have the backbone shortened by 4 carbons and are named tetranors. The advantages of measurements of these liver or kidney oxidation metabolites are their stability and credible estimation of the whole systemic production of parental compounds. An exception is LTE4, which is also secreted in urine and reflects the systemic production of CysLTs.
LIPID MEDIATORS IN SPECIFIC ASTHMA PHENOTYPES
Eicosanoids were studied in exercise-induced asthma because samples of EBC21 or urine showed increase in CysLTs related to the severity of symptoms following bronchoconstriction.22 This reflected bronchial mast cell activation by a non-immunological mechanism. There is also an increase in anti-inflammatory PGE2 and PGD2 systemic production after eucapnic hyperventilation-precipitating bronchoconstriction. This may explain the improved symptoms of exercise repeated within hours.20 It has been also shown that this phenotype depends on epithelial injury and increased epithelial 15-LO activity. Decreased lipoxin biosynthesis and isoprostane overproduction are also implicated in exercise-induced asthma.23
Asthmatic inflammation is a multicellular process involving both bronchial tissue and infiltrating inflammatory cells. In allergic asthma, immediate immunological reaction activates bronchial mast cells and results in bronchoconstriction, increased mucus secretion, edema, and cough. All these symptoms are evoked by 2 categories of mediators: preformed meditators, such as histamine, released by exocytosis and mediators produced by ongoing biosynthesis like eicosanoids. Contribution of each of these two mechanisms is complementary and variable. In clinics, neither inhibition of histamine receptors nor eicosanoid pathway effectively abolishes immediate reactions. The limited efficacy of leukotriene-modifying drugs inhibiting 5-LO activity or antagonizing CysLTR1 can be attributed to individual variability of these mechanisms and a high interindividual difference in leukotriene pathway activity. Urinary LTE4 can vary from less than hundred picograms per milligram creatinine in healthy subjects to several thousands in some asthmatic patients.24 There is also a debate whether CysLTs are produced by mast cells in the lung or by eosinophils. Recent studies on the eicosanoid handprint in different phenotypes of asthma showed that IS eicosanoid levels correlate well with peripheral blood eosinophilia and sputum eosinophil count.25 However, mast cells are also the abundant source of CysLTs. On the systemic level, excretion of urinary LTE4, an ultimate metabolite of LTC4 and LTD4, is elevated in mast cell disorders.26 Thus, both cells contribute to the production of these potent bronchoconstrictors in asthma. Mast cells also produce PGD2. This prostaglandin does not directly constrict bronchi through the DP1 receptor but stimulates sensory C-fibres to induce cough reflex.27 However, PGD2 can also activate the thromboxane receptor TP which is the least specific prostanoid receptor capable of contracting airway smooth muscle cells. Another biological effect of PGD2 is chemoattraction of eosinophils and innate lymphoid cells (ILC), a recently defined subset of innate immune cells. In particular, type-2 ILC (ILC-2) have the DP2 receptor (also named chemoattractant receptor expressed on Th2 cells, such as CRTH2 or GPR44).28 These cells are sources of IL-13 and IL-5 responsible for the persistence of allergic inflammation in the airways. Powell and Rokach29 discovered a novel eicosanoid which is another chemoattractant of eosinophils and more potent than PGD2. A product of 5-LO, 5-HETE, can be oxidized by a specific 5-HETE dehydrogenase to form 5-oxo-ETE. It was experimentally shown that 5-oxo-ETE activates and attracts eosinophils by a specific receptor named OXER1, previously cloned as an orphan receptor, GPCR48. Studies on 5-oxo-ETE and its receptor suggest that this eicosanoid can be responsible for eosinophilic inflammation of the airways. However, lack of the receptor homolog in mice is a major obstacle to investigate a conventional murine model of asthma. Using a feline model of allergic asthma in which cats were sensitized to Bermuda grass allergen, cells from BALF can produce 5-oxo-ETE.30 This lipid mediator induce chemotaxis of eosinophils at much lower concentrations than LTB4.
It remains unexplained why asthmatics are oversensitive to LTE4. Studies on the existence of a distinct LTE4 receptor overexpressed in asthma revealed some candidates. Nonspecific stimulation of adenosine receptors by LTE4 was proposed as the mechanism for a receptor cross-talk between CysLT1 and purinergic receptor P2Y12.31 However, subsequent studies did not support the explanation for enhanced asthmatic bronchial constriction in response to LTE4.32 Di Capite et al.33 proposed a mechanism in which CysLTs activation of mast cells mediated by the CysLTR1 receptor facilitate opening of calcium-release to activate calcium channels responsible for high sensitivity to this class of eicosanoid mediators in asthma.
It is frequently difficult to identify a single factor for chronic course of asthma in adults. Despite elevated total serum IgE, elevated Th2-type cytokines in the airways and allergic sensitization, asthma symptoms persist even after allergen avoidance. A protracted allergic inflammation is accompanied by induction of eicosanoid biosynthesis; however, no uniform pattern of these mediators can be identified.
Inflammatory phenotypes of asthma were proposed based on IS cellular content. However, prospective observational studies showed that around 40% patients changed their cellular phenotype of asthma during the observation period.34 This presentation of cellular inflammatory phenotypes can correlate with IS eicosanoid profiles only to some extent. However, if data on IS differential cell count were combined with the lipid mediator profile some interesting conclusions emerged.35 First, mild to moderate asthma can be distinguished from severe disease. The most distinct is the phenotype of severe eosinophilic asthma accompanied by chronic rhinosinusitis (CRS) in subjects with a low prevalence of atopy (26%). Most of them (86%) also manifest hypersensitivity to NSAIDs, and their asthma remains poorly controlled. These patients require high doses of inhaled corticosteroids, and 14% asthmatics need systemic corticosteroids. In this phenotype, eosinophilia is elevated both in peripheral blood and in IS. The profile of lipid mediators is dominated by high levels of LTD4, LTE4, and PGD2. This form of asthma represents classical AERD. However, there is another phenotype of severe asthma, without elevated eosinophil count in IS. The sputum is either neutrophilic or paucicellular, and two-thirds of asthmatics also have CRS and atopy is a common feature (65%). Asthmatics are mostly women, frequently overweight. More than one-third of these asthmatics have a history of asthma starting in childhood. Despite treatment with oral corticosteroids, these asthmatics have the worst control of the disease. In this specific phenotype, CysLTs or PGD2 is not elevated in IS, and the eicosanoid profile is marked only by a higher PGE2 concentration. It is noticeable that some of these patients also show hypersensitivity to NSAIDs. These 2 severe asthma phenotypes contrast with a mild to moderate asthma which is predominantly atopic and well controlled using low doses of ICS. None of these asthmatics have CRS, and IS shows no elevated eosinophil count or paucigranulocytic or neutrophilic one. None of these asthmatics have hypersensitivity to NSAIDs. In this form of asthma, no alterations in lipid mediators levels could be detected during a stable period of the disease. Among cellular and eicosanoid phenotypes of asthma, another mild to moderate form exists with a frequent CRS comorbidity, but without any other characteristic features of IS differential cell count or eicosanoid profile. These asthmatics rarely have atopy and their disease course is intermittent or moderate at most. It is surprising to observe sometimes NSAIDs hypersensitivity also within this group of asthmatics. Thus, sputum inflammatory cells match only partially with the profile of eicosanoids in the same samples. It seems, however, that both classifications correctly distinguish eosinophilic phenotypes. The major problem of phenotyping in asthma is classification of asthmatics with mixed or paucicellular sputum. It can be concluded, that a low content of eicosanoid lipid mediators, namely LTD4, LTE4, PGD2, and PGE2, predicts a favorable course of the disease, with good control of symptoms. This is not related to NSAIDs intolerance, atopy, or accompanying CRS. Using only urinary LTE4 excretion as a variable complementary to clinical characteristics of patients, similar subtypes of AERD can be distinguished.36
Hypersensitivity to NSAIDs like AERD has heterogenous presentations in asthmatics. Some important observations were recently published on the pathomechanism of this specific type of asthma. Liu et al.37 managed to test a murine model of the disease. It required to sensitize mice with a common house dust mite allergen and to ablate PGE2 production by microsomal PGE2 synthase knockout. A challenge with aspirin causes, in these animals, degranulation of mast cell and bronchoconstriction. There are post-challenge elevations in histamine and CysLTs. The same research group suggested that the major source of CysLTs in AERD patients could be transcellular biosynthesis by platelets adhering to peripheral blood granulocytes.38 Peripheral blood granulocytes of AERD asthmatics are also refractory to inhibition by PGE2.39 However, studies on nasal mucosa from AERD patients demonstrated another intriguing feature of the disease. Eosinophils infiltrating the upper respiratory tract not only produced CysLTs but also secreted interferon-γ.40 These mediators were absent in cells isolated from healthy controls or patients with chronic hyperplastic eosinophilic sinusitis as a disease control. Interferon-γ prestimulation of eosinophils was also necessary to activate CysLTs biosynthesis in eosinophils experimentally matured from the blood progenitors, and these cells also responded to ketorolac, another NSAID tested.41
Changes in the eicosanoid profile of IS immediately after positive bronchial provocation with lysine-aspirin are very interesting. These may help understand the initial reaction responsible for asthmatic attack in AERD patients. Biosynthesis of PGE2 in bronchi seems to be extremely sensitive to inhibition by acetylsalicylic acid.25 An inhaled lysine-aspirin dose of as low as 17 mg on average causes bronchoconstriction with an FEV1% decrease exceeding 20%. Interestingly, according to individual hypersensitivity to NSAIDs, smaller doses of the drug frequently elicit more severe contraction of bronchi. Despite a small total dose of the drug inhaled, a significant decrease in PGE2 sputum concentration is observed only in AERD. A much higher dose of acetylsalicylic acid inhaled by aspirin-tolerant asthmatics causes a decrease in the metabolite 13, 14-dihydro, 15-keto-PGE2,without PGE2 change. This 13, 14-dihydro, 15-keto-PGE2 is an intermediate metabolite of PGE2 inactivation pathway. It is surprising that in AERD patients, PGD2 is not rising after the drug provocation, although the prostaglandin remains elevated. Similarly, no changes in PGD2 metabolites are observed following inhalation of lysyl-aspirin. Thromboxane production is suppressed in aspirin-tolerant asthmatics receiving a full dose of lysine-aspirin, whereas no differences in the sputum levels of 11-dehydro-TXB2 are observed in AERD patients. Before the challenge, overproduction of CysLTs is manifested in AERD by higher LTE4, but concentrations of LTC4 and LTD4 in IS are comparable to those of aspirin-tolerant asthmatics. However, following a positive challenge in AERD group, a significant increase is noted for all CysLTs. The bronchial provocation does not affect the level of 5-HETE or 15-HETE in IS and only 12-HETE is decreased after provocation with acetylsalicylic acid, but this appears in both groups of asthmatics. An unexpected finding of the study is that overproduction of CysLTs does not correlate with the level of 5-HETE, a 5-LO pathway substrate. Following provocation with acetylsalicylic acid, LTB4 is the only eicosanoid mediator of 5-LO pathway which is decreased in AERD patients. Collectively, CysLTs overproduction occurs at the expense of LTB4 biosynthesis. The total 5-LO pathway eicosanoids show no changes after the provocation. It is highly plausible that following NSAIDs exposure, there is a dramatic shift in the profile of eicosanoids from LTB4 to CysLTs in the bronchi of AERD asthmatics due to enhanced transcellular biosynthesis. This might be explained by activation of platelets and their adherence to granulocytes. In fact, such an observation on elevated fraction of neutrophil-platelet aggregates in peripheral blood was described previously by Laidlaw et al.,38 demonstrating activated platelets in the granulocyte-infiltrated nasal mucosa. There remains, however, a pivotal question regarding the specificity of this finding and the reason for the enhanced aggregation between platelets and granulocytes in AERD. Results from allergen challenge (birch pollen) show no decrease in BALF LTB4 concentration. On the contrary, a trend toward increased LTB4 was accompanied by a robust response in 15-LO metabolites.42 Biosynthesis of CysLTs by the neutrophil-platelet aggregates seems a distinct feature of AERD. However, aspirin remains the most common antiplatelet cardioprotective drug administered. It irreversibly inhibits platelet COX-1 and decreases production of prothrombotic TXA2. Ex vivo experiments demonstrated the resistance of peripheral blood granulocytes from AERD subjects to PGE2 inhibition of transcellular CysLTs production and increased granulocyte-platelet aggregates.39 It is speculated that this resistance to anti-inflammatory activity of PGE2 might be related to altered EP receptor signal transduction and defects in 5-LO regulation by protein kinase A. However, we recently observed an inexplicable alteration in PGE2 biosynthesis during provocation of AERD patients with aspirin. This discovery may offer another explanation for the mechanism underlying the activation of platelets or even a direct constriction of bronchi in AERD.43 In EBC collected before and after inhalation challenge with lysine-aspirin, we identified an isomer of PGE2. While 8-iso-PGE2 is characterized by cisconformation of the 2 aliphatic chains at the cyclopentane ring, the regular PGE2 produced by a concerted action of cyclooxygenases and PGE synthases has trans-conformation. Thus, 8-iso-PGE2 is similar in its stereochemistry to 8-iso-PGF2α, the most commonly measured isoprostane product of arachidonic acid by non-enzymatic oxidation (Fig. 4). Also, 8-iso-PGE2 is a pro-inflammatory prostanoid, with broncho- and vasoconstrictory properties and has a similar activity as 8-iso-PGF2α.44 Following bronchoconstriction induced by the challenge test, we noticed a specific increase the EBC concentration of 8-iso-PGE2 only in AERD patients, whereas no changes in 8-iso-PGF2α were observed. This isoprostane production might be an initial event triggered by aspirin, activating platelet-neutrophil interaction and resulting in the release of CysLTs. Moreover, 8-iso-PGE2 can directly stimulate smooth muscle contraction. It would be necessary to investigate why this isoprostane is overproduced following exposure to NSAIDs in hypersensitive subjects. In vitro studies on macrophages stimulated with COX-2-inducing compounds, such as bacterial lipopolysaccharides (LPS), documented production of 8-iso-PGE2 within the first hours of the stimulation. We had similar observations on respiratory epithelial cells stimulated with LPS. During the induction of prostanoid biosynthesis, PGE2 accumulation was accompanied by a parallel increase in 8-iso-PGE2. Thus, 8-iso-PGE2 production occurs during induction of COX-2 enzyme, probably because the intracellular supply of arachidonic acid exceeds catalytic activity of cyclooxygenases. This is also due to inhibition of coexpressed COX-1 by NSAIDs. It requires a low basal level of COX-2, whose induction defect was recently described in fibroblasts of nasal polyps from AERD patients.45 This defect is attributed to a lowered density of the interleukin-1 receptor I and a failure to induce COX-2 and membrane-associated PGE synthase (mPGES). All these abnormalities can be corrected in vitro by artificial expression of EP2, the PGE2 receptor type-2, which has been shown to be associated with AERD in genetic studies. The appropriate cellular model of these eicosanoid anomalies may help prove the mechanism of NSAIDs-triggered bronchoconstriction in AERD. Respiratory epithelial cells collected from asthmatic patients tend to maintain their epigenetic marks for several passages. Recently, these primary cell cultures have been obtained by nasal brushing instead of bronchofiberoscopy.47 However, the fundamental question regarding the mechanism of dysregulation of COX-2, mPGES, and EP2 as a plausible cause for AERD will require more studies.
FUTURE DIRECTIONS IS THE RESEARCH ON EICOSANOID MEDIATORS IN ASTHMA
The last decade progress in our understanding of the role of eicosanoids in asthma have stimulated development of methods for screening and measurements of these lipid mediators in complex biological matrices. It is rather improbable that any single eicosanoid or even a complex profile of eicosanoids could provide explanation for the mechanism of asthmatic inflammation. It remains rather a practical question whether profiling of eicosanoids in clinical samples will offer any advantages over other diagnostic methods. One of the currently tested biomarkers is urinary LTE4 that promises a diagnostic validity in distinguishing AERD patients from aspirin-tolerant asthmatics. The sensitivity and specificity of this biomarker seem to be limited. From the clinical and practical points of view, a limited positive predictive value of urinary LTE4 precludes any safe recommendation for the tolerance of NSAIDs to a patient at risk for the disease. Another promising prediction based on urinary metabolites of eicosanoids is that systemic production of prostaglandins measured as a tetranor-PGE2 metabolite in urine, in children with wheezing, correlates with their risk for asthma. Systemic overproduction of PGE2 is associated with a suppressed ability of airway macrophages to phagocyte particulate matter carbon.46 A decreased number of the airway macrophage carbon particles was found in IS cytospin differential counts of asthmatic children. The impaired phagocytosis negatively correlates with disease severity as well as urinary excretion of 13, 14-dihydro-15-keto-tetranor-PGE2. Inhibition of macrophage phagocytosis by a micromolar concentration of PGE2, but not PGD2, was confirmed by in vivo experiments and is consistent with its anti-phlogistic activity. Ultimately, it might be expected that some rapid methods for assessment of eicosanoid mediators in EBC or IS will enable screening of asthmatics to better characterize their inflammatory phenotypes or even to monitor the course of the disease.2548 A recent metaanalysis on a diagnostic value of volatile organic compounds in the exhaled breath showed some encouraging results, although methods for eicosanoid detection and quantification in the exhaled breath still require collection of EBC, IS, or urine and laborious measurement techniques.49



 XML Download
XML Download