

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Air pollutants are believed to be a factor in the increasing prevalence of asthma and to exacerbate respiratory diseases.1 Diesel exhaust particles (DEPs) as a major component of fine particulate matter (PM2.5) in the atmosphere of urban areas can induce airway hyperresponsiveness (AHR) and inflammation by amplifying the T-helper 2 (Th2) immune response2345 and are associated with cardiorespiratory mortality and the aggravation of disease.678 DEPs can induce acute AHR and acute asthma exacerbations independent of their effects on allergic sensitization.9
Long-term exposure to air pollution causes chronic respiratory disease,10 and chronic inhalation of DEPs leads to the development of cough and sputum. In addition, chronic bronchitis may be partially responsible for some of the exacerbations of asthma.11 The biological response to inhaled particles is aggravated during chronic exposure to DEPs in a dose-dependent manner. In a previous study, inflammation and overproduction of mucus and surfactant components reached a plateau at 12 or 18 months of exposure during a 24-month experimental period, suggesting that DEPs play an important role in the development of chronic lung injury.12 Airway remodeling in asthma is defined by several structural changes, including epithelial cell mucus metaplasia, an increase in peribronchial smooth muscle mass, subepithelial fibrosis, and angiogenesis. Cytokines, chemokines, and growth factors released from inflammatory and structural cells in the airway are believed to play a pivotal role in the development of remodeling.13141516 Chronic inflammation is thought to initiate and perpetuate the cycles of tissue injury and repair in asthma, although remodeling may also occur in parallel with inflammation.17
Although long-term, repeated exposure to DEPs in air pollution increases the risk for chronic respiratory diseases and cardiorespiratory mortality,18 the biological mechanism and remodeling remain poorly understood during chronic exposure to DEPs. Therefore, we developed a mouse model of exposure to DEPs, and the effect of long-term exposure to DEPs as one of the air pollutants on airway inflammation and responsiveness, lung fibrosis, and goblet cell hyperplasia was examined.
Go to : 
MATERIALS AND METHODS
DEP sources and preparation
We obtained and used DEPs from Hajime Takizawa. DEPs were collected using the following method. The engine used for preparation of DEPs was a 4JB1 type (Isuzu Automatic Co., Tokyo, Japan), light-duty (2,740-cc), 4-cylinder diesel engine. The engine was connected to an EDGY dynamometer (Meiden-Sya, Tokyo, Japan) and operated using standard diesel fuel with a speed of 1,500 rpm under a load of 10 torque (kg/m). The exhaust was introduced into a stainless steel dilution tunnel (300×8,400 mm) in a constant-volume sampler system equipped to the end of the dilution tunnel. The temperature at the sampling point was below 50℃. The diameter of the particles was measured using an Anderson Air Sampler of the low-pressure type,19 and the mean diameter was 0.4 µm. Most of the shapes analyzed using a scanning electron microscope were globular. Details on the DEPs used were previously described.2021
Animal sensitization and exposure conditions
DEPs were sterilized by autoclaving and suspended in serumfree media after coating with BSA to minimize particle aggregation and hydrophobicity.22 After sonication, the endotoxin concentration of the DEP suspension was <0.064 ng/mL (0.32 EU/mL) using the Limulus Amebocyte Lysate assay (QCL-1000; BioWhittaker Inc., Walkersville, MD, USA).23 Female Balb/c mice, 5 to 6 weeks of age and free of mice-specific pathogens, were obtained from Orient Co, Ltd (Charles River Laboratories, Seoul, Korea). The mice were housed throughout the experiments in a laminar flow cabinet and maintained on standard laboratory chow ad libitum. All experimental animals used in this study were treated according to guidelines approved by the Institutional Animal Care and Use Committee of the Soonchunhyang University Medical School. DEPs were resuspended in saline solution for 30 minutes before administration. The mice (n=8 in each group) were exposed to 100 µg/m3 and 3 mg/m3 DEPs for 1 hour a day for 5 days a week from 4 to 12 weeks (Fig. 1) in a closed-system chamber attached to an ultrasonic nebulizer (NE-UO7; Omron Corporation, Tokyo, Japan) with an output of 1 mL/min and 1- to 5-µm particle size.
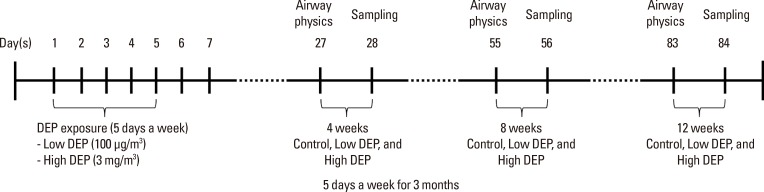
The control mice were administered and exposed to saline solution alone. Mice were sacrificed with an overdose of pentobarbital sodium (65 mg/kg body weight, administered intraperitoneally). The chest cavity was exposed, and the catheter was carefully inserted into the trachea and secured with ligatures. Bronchoalveolar lavage (BAL) was performed by 4 instillations of 1 mL of normal saline and gentle retrieval. Cell numbers were measured using a hemocytometer, and differential cell counts were performed on slides prepared by cyto-centrifugation and Diff-Quik staining (Scientific Products, Gibbstowne, NJ, USA). Supernatants were separated by centrifugation (500 g, 5 minutes) and maintained at -70℃ until use. After ligation of the right main bronchus, the left lung was fixed with 4% paraformaldehyde in phosphate-buffered saline and paraffin-embedded. The right lung was excised and immersed in TRI reagent (guanidinium thiocyanate-phenol mixture; Molecular Research Center Inc., Cincinnati, OH, USA), and immediately frozen in liquid nitrogen.
Determination of airway responsiveness to methacholine
Airway responsiveness was measured in unrestrained, conscious mice 1 day after the last challenge, as previously described.24 Mice were placed in a barometric plethysmographic chamber (All Medicus Co., Seoul, Korea), and baseline readings were taken and averaged for 3 minutes. Aerosolized methacholine in increasing concentrations (from 2.5 to 50 mg/mL was nebulized through an inlet of the main chamber for 3 minutes. Readings were taken and averaged for 3 minutes after each nebulization, at which time the enhanced pause (Penh) was determined. Penh, calculated as (expiratory time/relaxation time - 1)×(peak expiratory flow/peak inspiratory flow) according to the manufacturers' protocol, is a dimensionless value that represents the proportion of maximal expiratory to maximal inspiratory box pressure signals and the timing of expiration. Penh is used as a measure of airway responsiveness to methacholine. The results are expressed as the percentage increase of Penh following challenge with each concentration of methacholine, where the baseline Penh (after saline challenge) is expressed as 100%. Penh values averaged for 3 minutes after each nebulization were evaluated.
Preparation of lung tissues for histology and immunohistochemistry
Trachea and lung tissues were removed from the rats. Four percent paraformaldehyde fixing solution was infused into the lungs via the trachea. The specimens were dehydrated and embedded in paraffin. For histological examination, 4-µm sections of embedded tissue were cut on a rotary microtome, placed on glass slides, deparaffinized, and stained sequentially with hematoxylin and eosin.
Measurement of cytokine levels in BAL fluids
The cytokine levels were quantified in BAL fluid using a sandwich enzyme-linked immunosorbent assay kit according to the manufacturer's protocol (Biosource International Inc., Camarillo, CA, USA). Each sample was determined in triplicate. The lower limit of detection for interleukin (IL)-5, IL-13, interferon (IFN)-γ, IL-10, and vascular endothelial growth factor (VEGF) was 0.17, 30.00, 14.70, 15.60, and 2.00 pg/mL, respectively. Values below these limits were considered zero for statistical analysis. Inter- and intra-assay coefficients of variance were <10%.
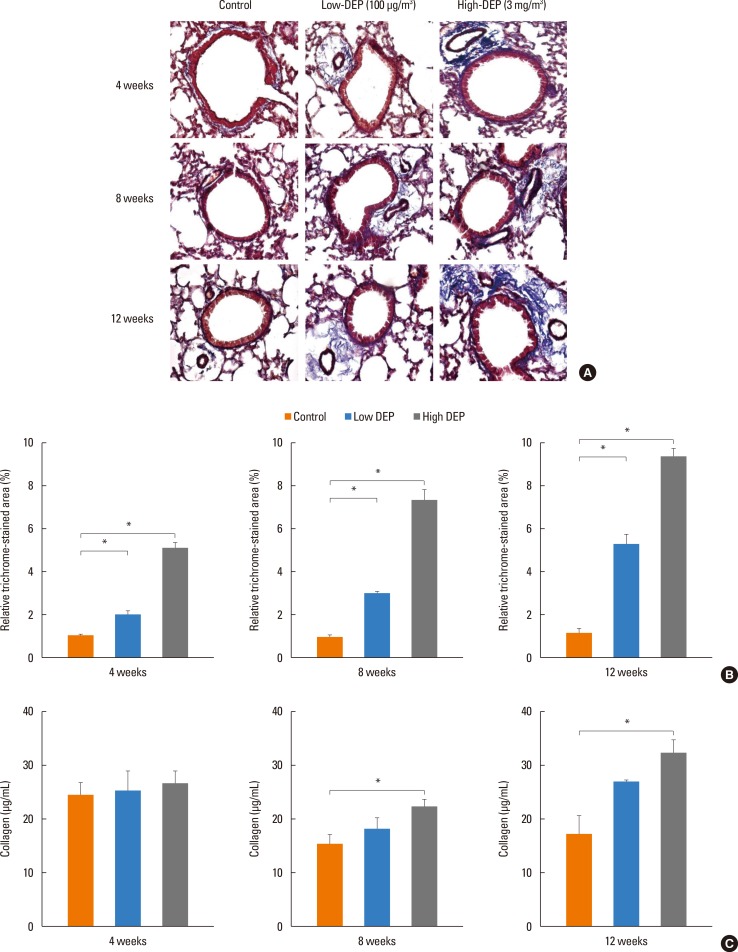
Collagen assay
Collagen assays were performed according to the user manual of the Sircol collagen assay kit (Biocolor, Northern Ireland, UK). Briefly, 100 µL of the protein extract sample in lung tissue was mixed with 1 mL of Sircol dye for 30 minutes and centrifuged at 10,000 rpm for 5 minutes to precipitate the formed collagen-dye complex. After decanting the suspension, droplets were dissolved in 1 mL of Sircol alkali reagent and vortexed. A total of 100 µl of the acquired solution was read at 540 nm.
Masson trichrome assay
The lung tissue slides were placed under a warmer at 60℃ for 30 minutes. Next, samples were immersed in xylene for 15 minutes, followed by 100%, 95%, 90%, 80%, and 70% ethanol for 10 minutes each. The slides were stained in accordance with the manual of the Masson trichrome assay kit (Sigma-Aldrich, St. Louis MO, USA). Briefly, Bouin's solution was used as a mordant to intensify the color reactions. The nucleus and cytoplasm were dyed using hematoxylin and scarlet acid. Phosphotungstic/phosphomolybdic acid solution was used to change a positive to a negative charge. After staining with aniline blue, the samples were treated with 1% acetic acid. Prior to observation, the samples were mounted with a cover slip using mounting media (Amresco, OH, USA). The positive trichrome-stained area was quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Values are reported as a percentage of positive areas within the total sample.
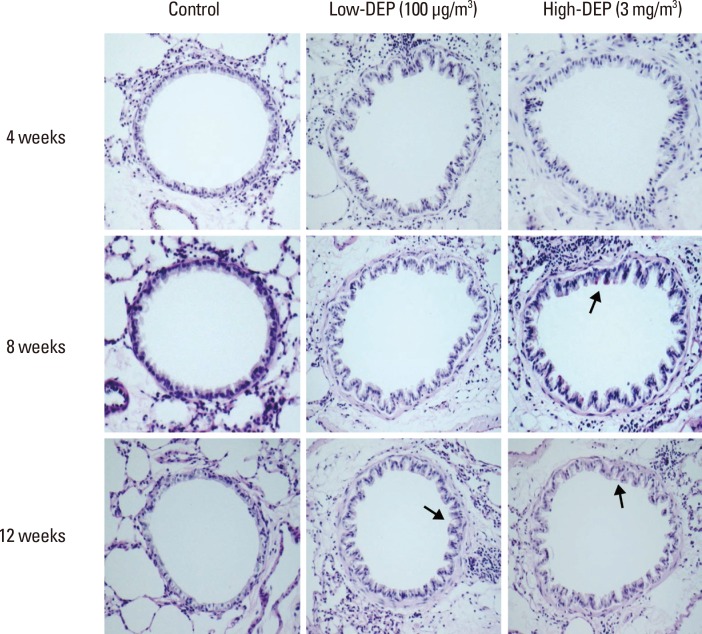
Periodic acid-Schiff assay
The slides were stained in accordance with the manual of a periodic acid-Schiff assay kit (Abcam, Cambridge, MA, USA). Briefly, lung tissue was cut into 4-µm sections and placed in periodic acid solution for 5 minutes. After washing with distilled water, the specimens were immersed in Schiff's fluid for 15 minutes. The slides were again washed with distilled water for 5 minutes and then placed in hematoxylin for 3 minutes. After the slides were exposed to running tap water for 3 minutes, bluing reagent was applied for 30 seconds. Before observation, the slides were mounted with a cover slip using mounting media (Amresco).
Statistical analysis
Differences between independent samples were compared using the nonparametric Kruskal-Wallis H test for continuous data. If differences were found to be significant, the Mann-Whitney U test was applied to compare differences between 2 samples. Differences were considered significant when a P value was <0.05. Results are expressed as mean±standard error of the mean (SEM) unless otherwise stated.
Go to :
RESULTS
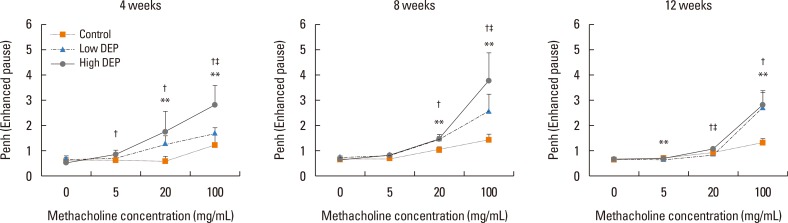
Airway responsiveness was measured 4, 8, and 12 weeks after the last exposure to DEPs in mice (Fig. 1). Airway responsiveness to aerosolized methacholine was measured in unrestrained, conscious mice. Mice were placed in the main chamber with a barometric plethysmograph and nebulized first with saline and then with increasing doses (from 2.5 to 100.0 mg/mL) of methacholine for 3 minutes for each nebulization. Readings of breathing parameters were taken for 3 minutes after each nebulization, during which time Penh values were determined. AHR was higher in the low-dose and high-dose DEP groups than in the control group and higher in the high-dose DEP group than in the low-dose DEP group at 4, 8, and 12 weeks (Fig. 2).
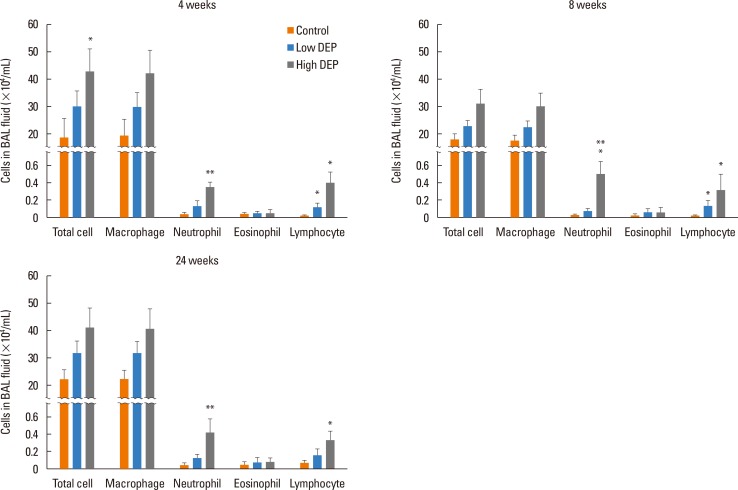
The total cell count in the BAL fluid tended to be higher in the low-dose DEP group than in the control group at 4, 8, and 12 weeks, and there was a significant increase in the total cell count in the high-dose DEP group than in the control group at 4 weeks (P<0.05) (Fig. 3). There were no differences in the number of macrophages among the 3 groups. The number of neutrophils was larger in the high-dose DEP group than in the control group at 4, 8, and 12 weeks (P<0.05 for each) (Fig. 3). The number of lymphocytes was larger in the high-dose DEP group than in the control group at 4, 8, and 12 weeks (P<0.05).
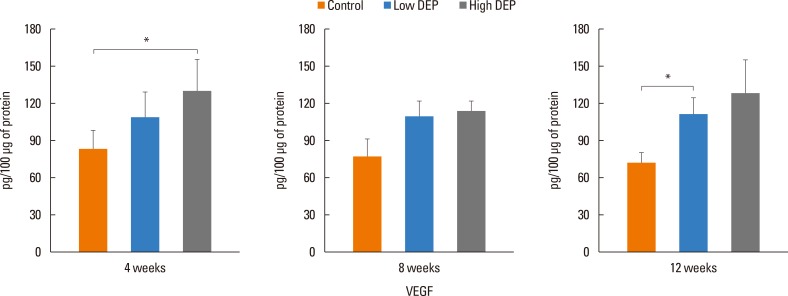
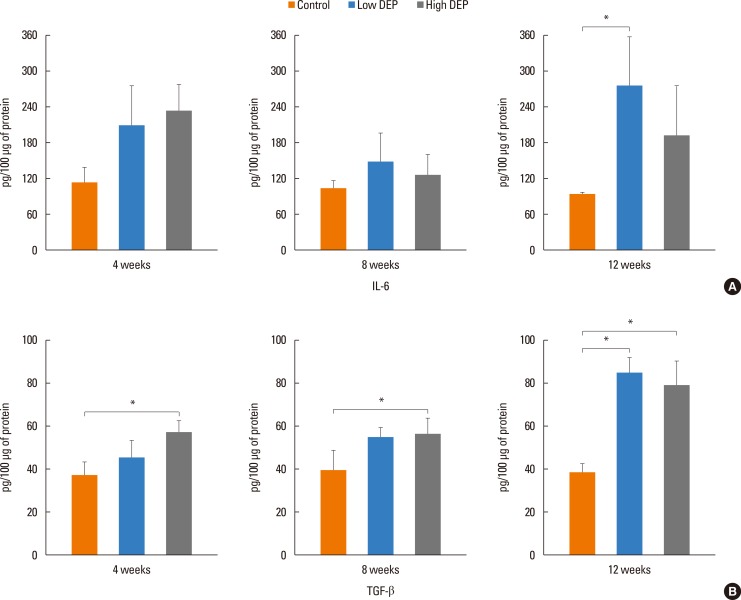
The level of IL-5 was higher in the low-dose DEP group than in the control group at 12 weeks (Fig. 4). The level of IL-13 was higher in the low- and high-dose DEP groups than in the control group at 12 weeks (Fig. 4). The level of IFN-γ was higher in the low-dose DEP group than in the control group at 12 weeks (Fig. 5). The level of IL-10 was higher in the high-dose DEP group than in the control group at 12 weeks (Fig. 6). The level of VEGF was higher in the low- and high-dose DEP groups than in the control group at 12 weeks (Fig. 7). The level of IL-6 was higher in the low-dose DEP group than in the control group at 12 weeks (Fig. 8A). The level of transforming growth factor-β (TGF-β) was higher in the high-dose DEP group than in the control group at 4, 8, and 12 weeks (Fig. 8B).
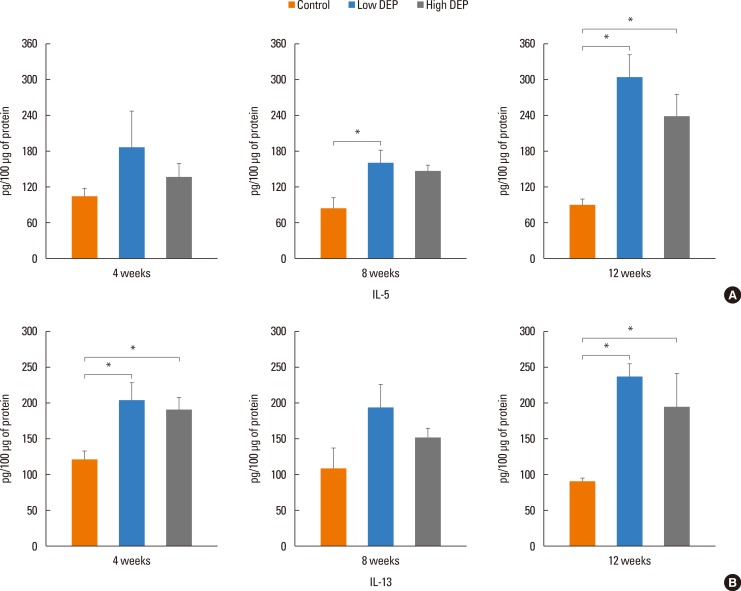 | Fig. 4Effect of DEPs on IL-5 and IL-13 levels in BAL fluid. Bars represent mean±SEM from the 6 independent experiments. *P<0.05 vs the sham group.
|
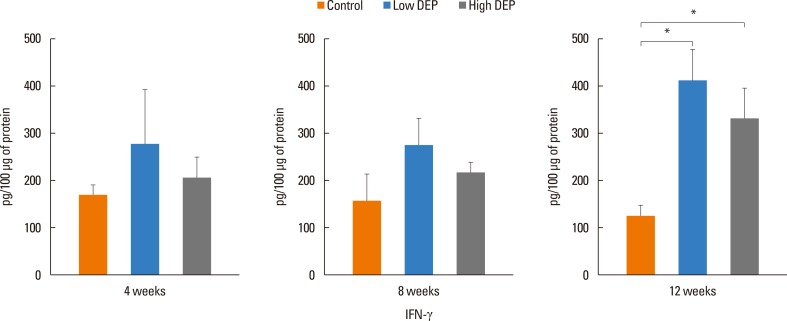 | Fig. 5Effect of DEP on IFN-γ level in BAL fluid. Bars represent mean±SEM from the 6 independent experiments. *P<0.05 vs the sham group.
|
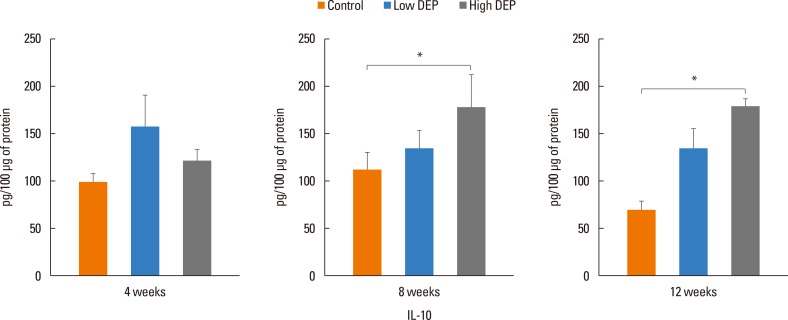 | Fig. 6Effect of DEP on IL-10 level in BAL fluid. Bars represent mean±SEM from the 6 independent experiments. *P<0.05 vs the sham group.
|
Lung fibrosis as shown by Masson trichrome staining of lung tissues was more severe in the low- and high-dose DEP groups than in the control group (Fig. 9A and B). The collagen content in lung tissues was greater in the high-dose DEP group than in the control group at 8 and 12 weeks (P<0.05) (Fig. 9C). Goblet cell hyperplasia in lung tissues was more severe in the high-dose DEP group than in the control group at 8 and 12 weeks (Fig. 10).
Go to :
DISCUSSION
This study revealed that long-term exposure to DEP leads to increased airway responsiveness and inflammatory cell infiltration, cytokine changes, collagen deposition, and goblet cell hyperplasia, indicating that long-term exposure to DEPs may be associated with AHR, inflammation, and lung fibrosis.
DEPs are the particulate component of diesel exhaust, which includes diesel soot and aerosols, such as ash particulates, metallic abrasion particles, sulfates, and silicates.25 Exposure to DEPs has been associated with acute short-term symptoms, such as headache, dizziness, light-headedness, nausea, coughing, difficult or labored breathing, chest tightness, and irritation of the eyes, nose, and throat.25 DEPs are related to allergic diseases, including asthma and allergic rhinitis based on extensive epidemiological studies252627 and act as an adjuvant during allergen exposure and effect acute asthma exacerbations, bronchitis, or chronic obstructive pulmonary disease.2829303132333435363738 Previous research has shown a strong link between particulate air pollution and detrimental health effects, including cardiopulmonary morbidity and mortality.39 In this study, AHR was higher in the low- and high-dose DEP groups than in the control group and higher in the high-dose DEP group than in the low-dose DEP group at 4, 8, and 12 weeks, suggesting that DEPs can induce AHR in a dose-dependent manner.
DEP exposure is associated with asthma and airway cell apoptosis. DEPs may directly contribute to asthma by inducing epithelial cell death through the apoptotic pathway40 and inducing inflammatory responses in human airway epithelial cells.41 DEPs activate intracellular signaling pathways that culminate in the production of profibrotic cytokines and growth factors.38 DEPs activate T cells in asthmatics with a higher effect during exacerbations, suggesting that uncontrolled asthma is a risk factor for aggravation in individuals exposed to traffic pollutants.42 DEPs also induce time-dependent increases in IL-8, granulocyte-macrophage colony-stimulating factor, and IL-1β in epithelial cell lines.42 DEPs affect dendritic cells (DCs), which results in increased production of TNF, IL-6, IFN-γ, IL-12, and VEGF.41 In costimulation assays of PM-exposed DCs and alloreactive CD4+ T cells, DEPs directed a Th2-like pattern of cytokine production (e.g., enhanced production of IL-13 and IL-18, and suppressed of production IFN-γ).43 Pro-oxidative DEP chemicals can interfere with Th1-promoting response pathways in a homogeneous DC population and provide a novel explanation for the adjuvant effect of DEPs on allergic inflammation, indicative of an adjuvant effect of particulate air pollutants in allergic inflammatory disease.44 DEPs induce antigen-independent DC maturation via epithelial cell-DC interactions mediated by the human bronchial epithelial cell-derived granulocyte-macrophage colony-stimulating factor.45 In our study, IL-6 and TGF-β were increased in the DEP long-term exposure group, suggesting that Th17 cytokine may be involved in airway inflammation following DEP exposure. Alveolar macrophages play an important role in particle-induced lung inflammation via direct induction of IL-13 production, suggesting that alveolar macrophages may act as major effectors of innate immunity to modulate immune and inflammatory responses toward a Th2-like condition via the production of IL-13, as observed in the adaptive immune response.46 In our study, IL-5, IL-13, INF-γ, and IL-10 were increased in the DEP long-term exposure group, suggesting that Th2, Th1, and regulatory cytokines are involved in airway inflammation following DEP exposure. In addition, DEP exposure has been associated with the upregulation of allergic immune responses and airway remodeling in both animal and human studies.4748 Long-term exposure can lead to serious chronic health problems, such as cardiovascular disease, cardiopulmonary disease, and lung cancer.35 Fibrotic reactions are a component of these pulmonary diseases and are involved in the progressive deposition of collagen by pulmonary fibroblasts.38 In this study, lung fibrosis, the collagen content, and VEGF in mice lungs exposed to long-term DEPs in a dose-dependent manner indicated that chronic exposure to DEPs can play a role in airway remodeling and angiogenesis. In addition, exposure to a higher dose of DEPs leads to increased airway inflammation, a higher cytokine response, and more severe lung fibrosis, suggesting that environmental reduction of DEP exposure is important for patients with airway diseases. In conclusion, chronic exposure to DEPs can cause AHR, inflammation, goblet cell hyperplasia, and lung fibrosis.
Go to :
 XML Download
XML Download