

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Chronic rhinosinusitis (CRS) is a heterogeneous disease characterized by local inflammation of the upper airways and sinuses. CRS is one of the most common chronic diseases in adults in the United States and affects up to 15% of the population.1234 Various theories on the etiology and pathogenesis of CRS, such as allergy, bacterial and fungal infections, and structural abnormalities, have been proposed, however, the pathogenesis remains poorly defined.5
CRS is typically classified into 2 types: CRSwNP and CRSsNP. Clinically, CRSwNP is associated more closely with clinical complaints of nasal obstruction and olfactory loss, and more frequently linked to comorbidities such as asthma and aspirin hypersensitivity. Immunologically, CRSwNP tissues are characterized by more intense eosinophilic infiltration and a Th2-based cytokine profile, whereas sinonasal tissues from CRSsNP have been reported to display a predominant infiltration of neutrophils and Th1 cytokines.6 However, there are still some controversies about the distinct role of Th1/Th2 profiles in the subtypes of CRS.
Dysregulation of the coagulation cascade may play an etiologic role in many diseases, including asthma, rheumatoid arthritis, glomerulonephritis, delayed-type hypersensitivity, and Crohn's disease, through excessive deposition of fibrin.78910111213 In the airways, there have been many reports on the relationship between asthma and dysregulated coagulation.78910111213 Furthermore, the link between asthma and CRS has been repeatedly observed as the concept of united airways disease (UAD).1415 Nonetheless, there have been very few reports on the role of the coagulation system in the pathogenesis of CRS. Shimizu et al.16 were the first to raise this issue by pointing out that thrombin activation occurs in nasal polyps. Recently, our group demonstrated excessive fibrin deposition caused by decreased fibrinolytic activity and increased expression of coagulation factors in the human nasal polyp (NP) tissue.1718 The goal of this review is to discuss the literature regarding the relationship between asthma and the coagulation cascade, and summarize our published and unpublished findings on dysregulation of the coagulation cascade as a contributor to the pathogenesis of CRSwNP.
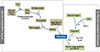
Coagulation and fibrinolytic cascades
Hemostasis, or the cessation of bleeding, occurs within the intravascular compartment lined with endothelium. Normal hemostasis and thrombosis involves a number of factors. Cellular factors include platelets, granulocytes, and monocytes and globular factors include coagulation (clot forming), the fibrinolytic (clot lysing) and anticoagulant (regulating) protein systems. Each of the three protein systems balances the activities of the other. Therefore, coagulation and fibrinolysis cascades describe the network of biological processes involved in fibrin clot manufacture and dissolution (Fig. 1).19
The initiator of physiologic hemostasis is factor VIIa which, when bound to its cofactor tissue factor (TF) activates factor X. Regulation of TF plays a major role in physiologic hemostasis. The activated prothrombinase complex, factors Xa and Va on platelets, then activates thrombin. Thrombin acts on the fibrin precursor protein fibrinogen to liberate the fibrin that forms the clot. Cleavage of fibrinogen by thrombin, along with the aggregation of platelets, can result in formation of a thrombus. The fibrin clot is formed by crosslinking of fibrin monomers and is further stabilized by FXIII, which is also activated by thrombin. Activated FXIII changes the formation of crosslinks in polymerized fibrin to yield the mature clot, and also crosslinks α2-plasmin inhibitor with fibrin to protect the newly formed fibrin from degradation by the fibrinolytic enzyme plasmin. The crosslinking of fibrin enhances its stiffness and rigidity, which allows it to retain plasma proteins. Thrombin in turn activates factor V, providing the shortest positive feedback loop to its own formation, because factor Va together with factor Xa activates thrombin. Antithrombin inactivates activated factor X and forms thrombin-antithrombin complexes, which irreversibly inhibit thrombin after about 15 minutes of coagulation. The anticoagulation system's role is to regulate all enzymes of the coagulation and fibrinolytic systems so that there is no inappropriate excess of clotting or bleeding.1920
Fibrinolysis is a process in which a fibrin clot, the product of coagulation, is broken down and prevents blood clots from growing and becoming problematic. The main fibrinolytic enzyme, plasmin, cuts the fibrin mesh at various places, leading to the production of fibrin degradation products (FDPs). Plasmin is produced from an inactive form, plasminogen, which is produced in the liver. Although plasminogen cannot cleave fibrin, it still has an affinity for it, and is incorporated into the clot when it is formed. Tissue plasminogen activator (t-PA) and urokinase plasminogen activator (u-PA) are the agents that convert plasminogen to the active plasmin, thus allowing fibrinolysis to occur. t-PA is released into the blood very slowly by the damaged endothelium of blood vessels, so that the clot is broken down after several days (when the bleeding has stopped). This is facilitated by the plasminogen entrapped within the clot; as it is slowly activated, it breaks down the fibrin mesh. t-PA and u-PA are inhibited by plasminogen activator inhibitor-1 (PAI-1). In contrast, plasmin further stimulates plasmin generation by producing more active forms of both t-PA and u-PA. Alpha 2-antiplasmin and alpha 2-macroglobulin inactivate plasmin by forming a 1:1 inhibitory complex with circulating plasmin. Plasmin activity is also reduced by thrombin-activatable fibrinolysis inhibitor, which modifies fibrin to make it more resistant to the t-PA-mediated plasminogen.1921
Asthma and the coagulation system
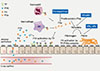
Coagulation and fibrinolysis were originally considered to be processes that take place in the vascular compartment. However, it is now appreciated that the airways represent a body compartment in which coagulation and anticoagulant mechanisms can be initiated and regulated locally.22 In addition to the activation of coagulation in lung inflammatory disorders that is probably induced by leakage of plasma proteins into the bronchoalveolar space, essential mediators of coagulation can be found locally in the lung, including TF that initiates coagulation, and thrombin, which transforms fibrinogen to fibrin.23 Several diseases associated with abundant lung inflammation, including acute respiratory distress syndrome, pneumonia, and lung fibrosis,2224 have been shown to result in similar changes in bronchoalveolar protein levels implicated in coagulation and fibrinolysis, suggesting that the physiologic equilibrium preventing fibrin clot formation swings toward a net procoagulant state. Particularly, this disturbed hemostatic balance in the airways may be important for the induction of allergic inflammation in asthma (Fig. 2), in which cytokines and protease-activated receptors (PARs) play an important role. In addition, platelets have been found to actively participate in many manifestations of asthma. Clinical evidence for a role of platelets in asthma is derived from studies demonstrating increased activation of platelets in patients with atopic asthma.2526 As well, increased circulating platelet-leukocyte aggregates have been detected in patients with asthma attacks and after allergen challenge.272829
As discussed above, cross-linked fibrin is the end product of coagulation and is generated after cleavage of fibrinogen by thrombin.23 Although fibrin is typically formed at sites of vascular injury, it can also be generated in the pulmonary compartment, wherein fibrin production is necessary for normal airway epithelial repair after epithelial damage.30 Severe asthma can be associated with exaggerated intra-alveolar fibrin production, as demonstrated by massive fibrin deposition in the alveoli and distal airways of an asthma patient who died from a severe asthma attack.31 Elevated concentrations of thrombin and thrombin-anti-thrombin complexes have been detected in sputum of patients with asthma3233 as well as in bronchoalveolar lavage (BAL) fluid after allergen challenge,3435 further supporting the existence of local coagulation activation in asthma. Thrombin activity in BAL fluid induced by segmental allergen provocation positively correlated with the degree of airway inflammation.35 In addition, the significance of increased coagulation activation in the lungs for the pathophysiology of asthma has been demonstrated in mouse studies. Exposure of mice to aerosolized fibrinogen followed by thrombin (which is expected to result in fibrin generation) caused increased airway hyperreactivity; thrombin or aerosolized fibrinogen alone were not sufficient to increase airway hyperreactivity to methacholine.31 Together, these data clearly indicate that elevated fibrin concentrations in the airways can produce a lung function disorder with some characteristics of asthma.
Thrombin has been implicated in asthma pathophysiology by both in vivo and in vitro studies. Administration of the thrombin inhibitor, PEG-hirudin, decreased airway hyper-responsiveness to methacholine in mice with acute allergic lung inflammation.31 Thrombin can mediate proinflammatory effects on a cellular level via cleavage and activation of protease-activated receptor 1 (PAR-1).36 Thrombin exposure stimulated mucin secretion by primary human bronchial epithelial cells; this effect could be mimicked by specific stimulation of PAR-1.37 In accordance with these in vitro observations, intranasally instilled thrombin induced hypertrophy of goblet cells in rat nasal epithelium.37 Relevant to asthma, thrombin-PAR-1 stimulation causes smooth muscle cell proliferation in vitro by stimulating platelet-derived-growth factor production and can induce connective tissue growth factor in fibroblasts,38 which is considered to contribute to development of fibrosis. In accordance, incubation of BAL fluid harvested from patients with atopic asthma challenged with allergen in a lung segment induced proliferation of fibroblasts in vitro, which could be inhibited by hirudin.34 In addition; thrombin can increase bronchial tone in human bronchial rings in vitro.39
Interventions targeting specific components of the coagulation system have been studied in a mouse model of allergic lung inflammation induced by ovalbumin challenge via the airways after prior sensitization. This model induces a clinical syndrome that at least in part resembles allergic asthma, characterized by eosinophilic lung inflammation, airway hyper-responsiveness, increased IgE levels, mucus hypersecretion, and eventually airway remodeling.40 Moreover, the similarity between the coagulation systems of mice and humans is considerable,41 making the mouse an attractive animal for studies on asthma and coagulation. Genetically modified FVIItTA/tTA mice with a very low expression of FVII demonstrated reduced coagulation activation in their lungs on ovalbumin challenge.42 Normal wild-type mice exposed to allergen showed increased FVII mRNA levels in whole lung homogenates and increased expression of FVII protein in bronchial epithelial cells, which was virtually absent in FVIItTA/tTA mice. Importantly, FVIItTA/tTA mice displayed a diminished influx of eosinophils into BAL fluid, together with lower levels of the chemoattractant eotaxin and the Th2 cytokines IL-4, IL-5, and IL-13. In addition, airway hyper-responsiveness and mucus layer thickness were reduced in allergen-challenged FVIItTA/tTA mice.42 FVIIa itself did not induce mucin production by human respiratory epithelial cells in vitro; however, addition of exogenous FX resulted in FXa production in these cell cultures, indicating the presence of functional TF/FVIIa, as well as enhanced mucin production.42 Addition of FXa to respiratory epithelial cells also induced mucin production.43 Together, these data suggest that TF/FVIIa mediated an effect on allergic lung inflammation, at least in part indirectly, via a role in the formation of FXa. In accordance with this hypothesis, FXa activity was found increased in BAL fluid of mice challenged with ovalbumin for 16 weeks (a model of chronic allergic lung inflammation associated with airway remodeling), concurrent with elevated FX mRNA levels in whole lung homogenates and alveolar macrophages.43 Treatment of mice with the FXa inhibitor, fondaparinux during the last 3 weeks of allergen challenge resulted in attenuation of airway hyperresponsiveness without altering infiltration of inflammatory cells into the lung and decreased the thickness of the mucosal layer and lung collagen deposition.43 The results of these studies introduce a novel participation of the coagulation system in the asthmatic response, indicating that coagulation plays an important role in experimentally induced allergic lung inflammation and that FXa/thrombin functions in airway remodeling by stimulating mucin and collagen deposition.
Some allergens, such as fungi, have proteolytic activity; however, there was no known relationship between allergen protease and TLR4 signaling until the recent report by Millien et al.13 They found that TLR4 has a critical role in the allergic response to fungal proteases, such as those derived from environmental aspergillus species. Colonization by aspergillus is frequently found in patients with severe asthma, and it causes allergic bronchopulmonary aspergillosis, a syndrome characterized by eosinophilic pulmonary infiltrates, high production of IgE, and central bronchiectasis. Millien et al.13 revealed that the development of airway inflammation, mucus hypersecretion, and bronchial hyperreactivity in response to the inhalation of an aspergillus protease depends on TLR4 and its downstream adaptor molecules. As compared with wild-type control mice, TLR4-deficient mice, on exposure to aspergillus protease, showed a scanty recruitment of type 2 innate lymphoid cells (despite normal numbers of Th2 cells) and a reduction in asthma-like features. They also found that the activation of TLR4 in wild-type mice by a fungal protease was indirect and required the presence of a serum factor; in their model, fungal proteases cleave the serum factor fibrinogen, thus releasing fibrinogen cleavage products (FCP) that can activate TLR4. Thrombin, the classic activator of coagulation, also generates FCP from fibrinogen, thus triggering TLR4. The thrombin inhibitor, hirudin was able to suppress aspergillus-driven asthma-like changes in mice, but it also suppressed asthma-like changes driven by the model antigen ovalbumin, which has no protease activity.13 Allergen challenge in humans and mice is generally accompanied by the extravasation of plasma, platelet aggregation, and activation of the coagulation cascade in the lung interstitium and bronchoalveolar compartment, with elevations of TF, thrombin, and fibrinogen, which probably explains how FCP can be generated with most allergens.11
In summary, plasma leak into the airways associated with activation of the coagulation cascade, predominantly FXa and thrombin, by endogenous or allergen-derived proteases by means of TLR4 signaling may contribute to allergic inflammation through downstream production of fibrin or by effects outside their role in hemostasis. Good examples of the latter are the increase of mucin production caused by FXa and the activation of PAR-1 on endothelial and epithelial cells by thrombin. This might be evidence that activation of the coagulation cascade in the airways doesn't simply reflect plasma leakage and following activation of clotting, but rather it can be more locally initiated. Administration of coagulation factors can reproduce some pathophysiologic alterations characteristic of asthma in animals in vivo and in relevant in vitro systems; conversely, inhibition of coagulation attenuates functional, immunologic, and morphologic features of allergic lung inflammation in mice elicited by ovalbumin sensitization and challenge.
In addition to a role for the activated coagulation cascade in the pathogenesis of asthma, the fibrinolysis system, especially PAI-1, has been implicated in collagen deposition and airway remodeling in the pathogenesis of asthma. We previously reported that levels of PAI-1 are elevated in the airways of a murine model of chronic asthma, and that PAI-1 deficiency is associated with reduced airway fibrosis in these mice.44 We also demonstrated that PAI-1 expression was elevated in the airways of patients with fatal asthma, and the 4G allele of PAI-1, which is associated with high plasma levels of PAI-1, was preferentially transmitted from parents with asthma to their asthmatic children.45 In addition, our data showed that elevated levels of plasma PAI-1 were associated with a decline of lung function in subjects with asthma.46 Other studies have also shown that PAI-1 levels in induced sputum samples from subjects with asthma were increased compared with healthy control subjects.4748 Furthermore, intra-airway administration of small interfering RNA against PAI-1 in mice attenuated not only airway hyperresponsiveness (AHR) and airway remodeling, but also the degree of eosinophilic airway inflammation in models of acute and chronic asthma.48 Recently, we reported the inhibitory effect of Tiplaxtinin, a PAI-1 inhibitor, on airway PAI-1 activity, lung inflammation, goblet cell hyperplasia, collagen deposition, and AHR in an OVA-challenged murine model of chronic asthma.49 Tiplaxtinin administration significantly reduced the inflammatory cell recruitment and the number of eosinophils in OVA-challenged mice. Airway tissue remodeling is thought to result from chronic repetitive injury to the airway wall caused by airway inflammation, and characterized by increased goblet cell hyperplasia. The number of goblet cells calculated by PAS staining was significantly lower in OVA-challenged mice with tiplaxtinin administration compared with OVA-challenged mice without tiplaxtinin treatment. Increased collagen deposition is a hallmark of airway remodeling due to prolonged inflammation in chronic asthma. Mice treated with tiplaxtinin during the challenge phase demonstrated significant inhibition of the collagen deposition compared with untreated OVA-challenged mice in Gomori trichrome staining. We further examined the physiologic effect of tiplaxtinin on AHR. The airway hyperreactivity of OVA-challenged mice was increased, as assessed by methacholine administration; however, this increased airway hyperreactivity to methacholine challenge in OVA-challenged mice was significantly reduced by tiplaxtinin administration. Taken altogether, these studies suggest that PAI-1 may play important roles in the pathogenesis of asthma by promoting airway inflammation, remodeling, and AHR, and PAI-1 may be a novel target of treatment of airway remodeling in asthma.
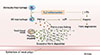
Excessive fibrin deposition in NPs caused by fibrinolytic impairment
Recently, we reported that excessive fibrin was deposited in NP tissue from patients with CRSwNP.17 The immunofluorescence analysis demonstrated significantly more intense fibrin staining in NP tissue from patients with CRSwNP than that seen in uncinate tissue (UT) from control subjects or patients with CRSsNP. Extravascular fibrin is ordinarily degraded to FDPs by plasmin to prevent excessive fibrin deposition.50 Therefore, we measured the levels of d-dimer, which is an important FDP, to evaluate the competence in fibrin degradation. Levels of d-dimer protein were significantly decreased in NP from patients with CRSwNP in comparison with levels in UT from patients with CRS or control subjects. Taken together, these findings suggest the presence of excessive fibrin deposition associated with reduced fibrin degradation in NP.
Fibrin degradation is facilitated by plasmin, which is generated through cleavage of plasminogen by u-PA and t-PA.51 We therefore assessed the expression of u-PA and t-PA in UT from patients with CRSsNP or CRSwNP and from control subjects as well as in NP from patients with CRSwNP. Although the expression of mRNA for u-PA was not different among the four groups, t-PA mRNA levels were significantly decreased in NP tissues from patients with CRSwNP compared with UT from patients with CRS or control subjects. To confirm this observation at the protein level, we measured the concentration of u-PA and t-PA in homogenates of UT and NP tissues by ELISA. The results were in accordance with the mRNA data; t-PA protein levels were significantly decreased in NP from patients with CRSwNP compared with UT from patients with CRS or control subjects, however, u-PA protein levels were not different among the four groups. Together, these results show a reduction of t-PA mRNA and protein levels, and suggest that the fibrinolytic pathway is compromised in NP tissue and may play a role in the excess deposition of fibrin in polyps.17
NPs from patients with CRSwNP have long been known to be characterized by Th2-dominant eosinophilic inflammation.52 In order to verify whether levels of plasminogen activators correlated with eosinophilic inflammation in nasal tissues, we assayed the levels of ECP as a marker for the presence of eosinophils in nasal tissue. The concentration of t-PA in UT and NP was significantly negatively correlated with the concentration of ECP.17 However, the concentration of u-PA in nasal tissue did not show a significant correlation with the concentration of ECP. In immunohistochemical studies, t-PA expression was observed in epithelial cells. Given that expression of t-PA was reduced in NP tissue and negatively correlated with ECP, we hypothesized that Th2 cytokines might regulate t-PA expression in airway epithelial cells. To test this, primary normal human bronchial epithelial cells (NHBE) and nasal epithelial cells (NEC) were stimulated with Th2 cytokines, IL-4, or IL-13 for 24 hours. The levels of t-PA mRNA, but not u-PA mRNA were significantly down-regulated by both Th2 cytokines in a dose-dependent manner. To confirm this observation at the protein level, we measured the concentration of plasminogen activators from cell lysate of NHBE cells using ELISA. Although the levels of u-PA protein were not altered by Th2 cytokine stimulation, the levels of t-PA protein were significantly down-regulated by both Th2 cytokines. These results suggest that Th2 cytokines down-regulate expression of t-PA, but not u-PA in airway epithelial cells.17
These results suggest a model in which vessel permeability is increased in inflamed sinus tissues, resulting in the leakage of plasma proteins into the extravascular compartment. Much of the extravagated fibrinogen can be rapidly converted to fibrin. In inflamed tissue of CRSwNP patients, down-regulation of t-PA by Th2 cytokines leads to insufficient fibrin degradation, resulting in excessive fibrin deposition in NP tissue, a factor that might contribute to polyp growth (Fig. 3). In addition to the tissue remodeling actions of fibrin deposition, the proinflammatory effects of excessive deposition of fibrin may play an etiologic role in NP (see above). This model suggests that prevention of fibrin deposition may be a potential new strategy for novel therapeutic approaches to CRSwNP.17
Increased expression of coagulation factor XIII-A in NPs
We analyzed data from a previously performed microarray analysis to compare coagulation factor gene expression in UT from patients with CRSsNP and CRSwNP, and control subjects, as well as in NP tissue from patients with CRSwNP.53 There was a substantial increase in mRNA levels of the A subunit of coagulation factor XIII (FXIII-A) in NP tissues of CRSwNP patients, compared with levels seen in UT from either patients with CRS or control subjects. Coagulation factor XIII (FXIII) is a transglutaminase that participates in the final stage of the coagulation cascade, crosslinking of fibrin. There are plasma and cellular forms of FXIII. Plasma FXIII has 2 enzymatically active A subunits (FXIII-A) and 2 inhibitory/carrier B subunits (FXIII-B) that form a tight tetrameric complex (FXIIIA2B2), whereas cellular FXIII is a dimer of FXIII-A, present in platelets, monocytes, and macrophages.5455 In contrast to FXIII-A, our data showed no difference in UT mRNA levels of FXIII-B between patients with CRS and control subjects.
For further confirmation of microarray data, we performed Real-Time PCR to assess the expression of FXIII-A in UT from patients with CRSsNP and CRSwNP, and control subjects, as well as in NP tissue from patients with CRSwNP. FXIII-A mRNA levels were significantly increased in NP tissues from patients with CRSwNP compared with levels in UT from either patients with CRS or control subjects. In order to confirm the protein level, we measured the concentration of FXIII-A in homogenates of UT and NP tissues by ELISA. In agreement with the mRNA data, FXIII-A protein levels were significantly increased in NP tissue from patients with CRSwNP in comparison with those seen in UT from either patients with CRS or control subjects. In addition, immunohistochemical analysis of surgical samples from control subjects and patients with CRS revealed FXIII-A+ signals mainly in submucosal inflammatory cells. We counted the number of FXIII-A+ inflammatory cells and confirmed that FXIII-A+ inflammatory cell numbers were significantly increased in NP from patients with CRSwNP, compared with those seen in UT from either patients with CRS or control subjects.
FXIII-A is expressed primarily in platelets, megakaryocytes, and macrophages.56575859 We therefore performed dual immunofluorescence analysis by using anti-FXIII-A and antibody against a marker of macrophages (CD68). We found a high degree of colocalization of FXIII-A with CD68+ macrophages in NP. Macrophages are widely recognized to be polarized by their microenvironment, especially by T-helper cytokines and pathogens.6061626364 Classical activated macrophages (also known as M1 macrophages) develop in response to proinflammatory stimuli, such as Th1 cytokines (IFN-γ) or bacterial products (LPS). In contrast, alternatively activated macrophages are induced by exposure to Th2 cytokines, including IL-4 and IL-13, and are therefore called M2 macrophages. Recent studies have shown that expression of FXIII-A is increased in M2 macrophages.5965 So, we next examined whether M2 macrophages are a major source of FXIII-A production in NP. We first determined expression levels of M2 macrophage markers, macrophage mannose receptor (MMR), CD163, and stabilin 1 (STAB1) in UT and NP using Real-Time PCR. Levels of mRNA for MMR, CD163, and STAB1 were significantly upregulated in NP, compared with those seen in UT from either patients with CRS or control subjects. We also found that the expression of FXIII-A significantly positively correlated with the expression of MMR, CD163, and STAB1. To further investigate whether M2 macrophages were the FXIII-A-producing cells in NP, we performed triple-immunofluorescence analysis by using anti-FXIII-A, CD68, and CD163. We detected FXIII-A+ signals in CD68+ and CD163+ cells in NP. These results suggest that M2 macrophages are the sole or major FXIII-A-producing cells in NP.
In summary, the tissue level of FXIII-A was profoundly increased in NP tissue and M2 macrophages are the major source of FXIII-A in NP. Overproduction of FXIII-A may lead to the acceleration of the coagulation cascade, resulting in excessive fibrin deposition, which, in turn, retains exuded plasma proteins, and participates in tissue remodeling, intense edema, or pseudocyst formation in the submucosa of NP tissue (Fig. 4). Our results imply that targeting the local production of FXIII-A from M2 macrophage might be of therapeutic value for treating patients with CRSwNP.18
Animal model systems are valuable for investigating human diseases. Our laboratory recently established a murine model of NP using a protocol generated by Kim et al.66 and investigated similarities and differences between this murine model and human NP. Similar to human NP data that we have published, we could detect markedly increased deposition of fibrin and significant elevations of gene and protein expressions of coagulation factors in the nasal mucosa of the NP mouse model compared to control (unpublished data). These preliminary data demonstrated that the NP mouse model showed enhancement of fibrin deposition and the coagulation cascade, reminiscent of human NP. This mouse model may enhance our understanding of the pathophysiology of NP and provide a way to test coagulation and fibrinolytic systems for their causative role and therapeutic application.
Role of thrombin in chronic rhinosinusitis-associated tissue remodeling
Shimizu et al.16 reported the first study to show the presence of thrombin and thrombin-antithrombin (TAT) complex in nasal secretions, and that thrombin and TAT were significantly increased in nasal secretion of patients with AR and CRSwNP with asthma compared with the control group. They also revealed that thrombin and protease-activated receptor (PAR)-1 agonist peptides significantly stimulated the secretion of vascular endothelial growth factor (VEGF) from NHBE cells and NEC. Taken together, these results suggest that significant local activation of the coagulation system occurs in the upper airways of patients with CRSwNP, and that its effector enzyme thrombin promotes tissue remodeling by stimulating the secretion of VEGF from airway epithelial cells.16
Shimizu et al. also found that thrombin and TNF-α enhance TF activity on the surface of airway epithelial cells. TF is a transmembrane glycoprotein that is an initiating protein and the most potent stimulator of the extrinsic coagulation cascade. TF is expressed in airway epithelial cells including NEC.37 Factor VIIa (FVIIa) binds to TF on the cell surface and this complex binds to factor X (FX), converting it to the activated form factor Xa (FXa), which leads to eventual thrombin (FIIa) formation and fibrin deposition. Therefore, their finding suggests that thrombin combined with TNF-α can lead to continuous activation of the coagulation system in upper airway inflammation. Thrombin can also promote inflammatory responses by stimulating the production of inflammatory cytokines and growth factors, including IL-6, IL-8, PGE2, CCL2, platelet-derived growth factor (PDGF), and the mucin MUC5AC from airway epithelial cells, and via these mediators it can affect airway permeability and eosinophil migration.37386768697071
In summary, activation of the coagulation system with increased thrombin generation is involved in the pathogenesis of tissue remodeling in the upper airways. This observation suggests that mucosal dysregulation of the coagulation system in the nose may be one feature of CRS that should be further explored to establish its pathogenic role.16
CONCLUSIONS
As enhanced coagulation and dysregulated fibrinolysis have been well established to be prominent pathogenic features of asthma, the role of dysregulated coagulation in CRS with NP, which is highly comorbid with asthma and shares several other pathogenic mechanisms, is worthy of further investigation. Disorders in the coagulation and fibrinolytic systems have been implicated in CRS by demonstration of thrombin activation, excessive fibrin deposition, decreased t-PA and increased expression of coagulation factor XIII-A in NP tissue.161718 In addition, the NP mouse model that has recently been established also showed enhanced activity of the coagulation cascade and fibrin deposition, as occurs in human NP. Taken together, focus on the coagulation and fibrinolysis systems may advance the understanding of pathophysiology of NP, and provide alternative and novel therapeutic modalities for CRSwNP, such as anticoagulants or fibrinolytic agents.

 XML Download
XML Download