

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
With acceptance of the concept that asthma is a heterogeneous disease, several recent studies are now confirming that severe asthma can present in multiple different ways from traditional asthma. Although the term "severe asthma" remains vague, severe asthma includes existing disease entities: steroid-insensitive asthma, steroid-resistant asthma, and refractory asthma. These subsets of asthmatics seem to be estimated up to 5%-10% of all asthmatics.1 Since current treatment guidelines for asthma are based on the typical phenotypic group of T helper type 2 (Th2)-predominant asthma, the major unmet needs of current therapies comprise better treatment of patients with severe asthma. In fact, large scales of studies have identified several phenotypes of severe asthma.2 However, these phenotypes do not identify the immunopathology that makes these phenotypes distinct or determine the suitable group for a specific or tailored therapeutic approach. With this trend, the term "endotype" has emerged.2 Thanks to many studies for a better understanding of endotypes for asthma, several novel targets have been suggested to be implicated in the pathogenesis of bronchial asthma, especially severe asthma. Although many characteristics have been released from each endotype of asthma, severe asthma has steroid resistance, a common characteristic. Thus, we cannot take our attention off steroid resistance to understand the mechanisms of severe asthma. To date, several molecular mechanisms of glucocorticoid resistance have been elucidated in asthmatic patients. These are genetic association, transcriptional regulation, suppression of histone deacetylation, and immune mechanisms. Our recent work has shown that endoplasmic reticulum (ER) stress is importantly involved in the pathogenesis of bronchial asthma, especially steroid-resistant bronchial asthma at least in part through modulation of nuclear factor-κB (NF-κB).3 Interestingly, the results have indicated that development of ER stress and activation of unfolded protein response (UPR) in airway epithelial cells as well as inflammatory cells are critical in the induction and maintenance of severe asthma.
In this review, we describe the roles of ER stress in severe steroid-resistant asthma and its potential therapeutic applications to surpassing current medications targeting traditional Th2 cell-mediated allergic inflammation.
Severe asthma and steroid resistance
For many years, the term "severe asthma" has been used interchangeably with other similar terms, and considerable effort has been concentrated to establish the term and concept. A World Health Organization (WHO) meeting has suggested that severe asthma includes 3 groups, each carrying different public health messages and challenges: (1) untreated severe asthma, (2) difficult-to-treat severe asthma, and (3) treatment-resistant severe asthma.4 To date, although severe asthma has also been recognized as a heterogeneous disorder, it is usually defined as failure to achieve control with maximum doses of inhaled corticosteroid therapies,1 which is consistent with the last group, treatment-resistant severe asthma suggested by WHO meeting. Taken together, severe asthma contains existing disease entities: steroid-insensitive asthma, steroid-resistant asthma, and refractory asthma. These subsets of asthmatics have been estimated up to 5%-10% of all asthmatics.
Glucocorticoids are the mainstay of anti-inflammatory therapy in respiratory disorders, including bronchial asthma.5 In fact, most of the asthmatic patients can be controlled by low doses of inhaled corticosteroids (ICSs), which have become first-line medication for patients with asthma, as recommended in several international guidelines for the management for bronchial asthma. Despite this proven clinical efficacy of glucocorticoids, about 10% of patients with bronchial asthma require the maximal dose of ICSs, and approximately 1% of patients are controlled only by oral administration with glucocorticoids. Rarely, some patients are completely refractory to glucocorticoids, even high doses of oral glucocorticoids. This entity of patients is clinically called as steroid-resistant asthmatics, as defined by no improvement after high doses of an oral corticosteroid (40 mg/day prednisone for 2 weeks).6 As mentioned above, severe asthma means a condition of no or less response to appropriate treatment under a correct diagnosis of bronchial asthma. Therefore, steroid resistance may be a mechanism contributing to asthma severity.7 Although it remains a very difficult problem to quantify steroid resistance, several molecular mechanisms for steroid action and resistance are now better understood, and this issue has identified new targets for the treatment of severe asthma.
Molecular mechanisms of steroid resistance
Understanding the anti-inflammatory action mechanisms of corticosteroids is required to comprehend the molecular basis of steroid resistance by priority. Therefore, we want to have an overview of action mechanisms for the anti-inflammatory effects of corticosteroids in asthma prior to explore the mechanisms leading to steroid resistance.
Molecular action mechanisms of glucocorticoids
The anti-inflammatory molecular action mechanisms of corticosteroids can be classified into 2 groups: (1) genomic mechanisms which are the increased expression of anti-inflammatory genes and the decreased expression of proinflammatory genes and (2) non-genomic mechanisms.
Genomic mechanisms: anti-inflammatory gene transactivation
When glucocorticoid binds to the glucocorticoid receptor (GR), GR dissociates from its cytoplasmic complex and is translocated into the nucleus. In the nucleus, the glucocorticoid-GR binds to glucocorticoid response elements (GREs), thereby increasing transcription of glucocorticoid response anti-inflammatory genes, such as mitogen-activated protein (MAP) kinase phosphatase 1 (MKP-1), and glucocorticoid-inducible leucine zipper 1 (GILZ-1).8
Genomic mechanisms: proinflammatory gene suppression
The major action of corticosteroids in suppression of inflammation is to switch off the activated multiple genes that encode for various inflammatory mediators and receptors.9 At first, it was seen as a dominant mechanism for proinflammatory gene suppression, that is, glucocorticoid binds to DNA-bound proinflammatory transcription factors, such as NF-κB and activator protein-1 (AP-1). In fact, while this direct transrepression mechanism is now known to have weaker and less specific significance than before, the recruitment of histone deacetylase 2 (HDAC2) to the activated inflammatory gene complex by ligand-bound GR with suppression of multiple activated inflammatory genes has been shown to account for the powerful effects of glucocorticoid in controlling inflammation.10
Non-genomic mechanisms
Glucocorticoid can increase the production of nitric oxide (NO) that displays multiple functions, including anti-inflammatory property.11 It can also induce the phosphorylation and extracellular release of annexin-1.12 There is recent evidence for an important role of extracellular annexin-1 as an inflammation-resolving protein, acting on the formyl peptide receptors FPR1 and FPR2.13
Molecular mechanisms of glucocorticoid resistance
Genetic association
Several studies have suggested that genetic factors can affect responsiveness for corticosteroids.14,15 For example, the bone morphogenetic protein receptor type II (BMPR II), one of the genes showing differential expression according to steroid responsiveness in humans, has enhanced steroid responsiveness when transfected into cells. A functional polymorphism of the gene glucocorticoid-induced transcript 1 (GLCCI1) linked to corticosteroid-induced apoptosis is suggested to be associated with responsiveness to ICSs in asthmatic patients. To date, however, there is no evidence for the link between polymorphisms/structural abnormalities in GR and steroid resistance in asthmatic patients, although a polymorphism of GRβ is associated with a reduced response to corticosteroids.16 In addition, some studies have demonstrated that the MDR1 gene, which encodes P-glycoprotein, is up-regulated in inflammatory cells from patients with glucocorticoid-resistant inflammatory bowel disease and rheumatoid arthritis17,18; however, the expression of P-glycoprotein has been shown to correlate with increased intracellular dexamethasone levels in airway epithelial cells, suggesting that up-regulation of P-glycoprotein is unlikely to be a mechanism of steroid resistance in lung epithelium.
Functional changes in GRs
GR phosphorylation by several kinases is an important component for the reduction in GR function through altering its binding stability, translocation into the nucleus, binding to DNA, and interaction with other proteins, including transcription factors and molecular chaperones.19 Exposure to cytokines, such as IL-2, IL-4, and IL-13, and subsequent activation of mitogen-activated protein kinase (MAPK) have been found to induce glucocorticoid resistance in inflammatory cells through inhibition of GR ligand binding, and the effect is suppressed by a p38 MAPK inhibitor.20 In fact, a p38 MAPK inhibitor inhibits phosphorylation of serine 226 (Ser226) on GR, which is induced by IL-2 and IL-4, and this effect is mostly observed in peripheral blood mononuclear cells (PBMCs) from asthmatics.21 IL-2 has been shown to inhibit GR nuclear translocation through p38 MAPK signaling with STAT5.22 Also, c-Jun N-terminal kinase (JNK) can directly phosphorylate GR at Ser226, resulting in inhibition of GR binding.23 MKP-1, an endogenous inhibitor of p38 MAPK and JNK signaling, is activated by corticosteroids. In alveolar macrophages obtained from severe asthmatic patients with reduced MKP-1 expression as well as murine macrophages from MKP1 knockout mice, reduced steroid responsiveness has been found.24 Moreover, there is a close correlation between reduced steroid-induced MKP-1 expression and increased p38 MAPK activity.24 In addition, the serine/threonine phosphatase protein phosphatase 2A (PP2A) is involved in the dephosphorylation of phosphorylated GR.25 Supporting this contention, PP2A expression and activity are reduced in PBMCs from patients with steroid resistance, and knockdown of PP2A or okadaic acid, an inhibitor, reduces steroid responsiveness and GR Ser226 dephosphorylation with nuclear translocation as well as an increase in JNK1 phosphorylation.
Microbial origin stimuli can induce steroid resistance in airway inflammatory cells. Staphylococcal enterotoxin B induces steroid resistance in human T cells in vitro through activation of the extracellular signal-regulated kinase (ERK) pathway linked to GR phosphorylation.26 Additionally, interferon (IFN)-γ inhibits GR nuclear translocation through activation of the TLR4/MyD88 pathway in murine pulmonary macrophages.27 TLR4 signaling has also been shown to contribute to stress-induced splenic glucocorticoid resistance in mice.28 Furthermore, recent reports have demonstrated that activation of TLR7 and TLR9 induces steroid resistance in plasmacytoid dendritic cells (DCs) from patients with systemic lupus erythematosus (SLE) and 2 lupus-prone animal models.29
GR can be nitrosylated by NO donors, resulting in reduced binding affinity for corticosteroids.30 It has been well known that patients with severe asthma produce high levels of NO, which nitrosylate the GR at the HSP90 binding site, resulting in a decrease in the affinity of GR to glucocorticoid as well as HSP90.20 However, the relevance of defective GR nuclear translocation and binding affinity in structural cells has not been limited, although airway epithelial cells have been accepted as an important immune/inflammatory responder.
GR isoform identity and expression
GRα predominates in most cell types but other isoforms do arise as a consequence of alternative splicing, and responsiveness to glucocorticoids can be modulated by the relative levels of the expression of each GR isoform.31 GRβ has been known to act as a dominant negative inhibitor through various mechanisms, including binding to GRE, formation of a heterodimer with GRα , interruption of nuclear translocation of GRα , interaction with transcriptional factors.32 Exposure to cytokines increases the expression of GRβ in airway epithelial cells and various inflammatory cells.33 Moreover, TGF-β1, which is known to be associated with asthmatic airway remodeling, has been shown to reduce glucocorticoid responses in A549 cells, partly due to decreased GRα expression.34
Activation of proinflammatory transcription factors
In inflammatory cells, the transcription factors NF-κB, STAT5, and AP-1 have been implicated in the occurrence of steroid resistance. Among them, AP-1, a heterodimer of Fos and Jun protein, may be the most important transcriptional factor associated with steroid resistance of asthma because AP-1 physically interacts with GR, thereby preventing its binding to GREs and other transcriptional factors,35 which is supported by high levels of AP-1, phosphorylated JNK, and c-Fos observed in inflammatory cells from glucocorticoid-resistant asthmatics.35 NF-κB activation is correlated inversely with glucocorticoid responsiveness in patients with severe asthma, and STAT5 is known to be implicated in defective GR nuclear translocation and DNA binding in HT-2 cells.22 Meanwhile, although it remains to be established whether transcriptional factors are increased in the context of structural cells including epithelial cells, activation of IRF-1 by IFNs or TNF-α may contribute to steroid resistance in airway smooth muscle cells.36
Defective histone acetylation
Recruitment of HDAC2 to activated inflammatory genes is a major mechanism of inflammatory gene repression by glucocorticoids; expression of HDAC2 is reduced in some diseases in which patients respond to steroids poorly.37 There is strong evidence connecting decreased HDAC2 activity with steroid resistance; molecular mechanisms for decrease HDAC2 expression/activity have recently been elucidated.38 In addition, low levels of HDAC2 expression have been reported in PBMCs and alveolar macrophages from refractory asthmatics and in airways of smoking asthmatics.39,40 Both oxidative and nitrative stresses play a crucial role in reducing HDAC2 expression/activity, which enhances formation of peroxynite that nitrates tyrosine residues of HDAC2, thereby leading to its inactivation, ubiquitination, and degradation.41 In particular, phosphoinositide 3-kinase (PI3K)-δ activated by oxidative stress is implicated in the phosphorylation and inactivation of HDAC2.42 In lung epithelial cells, cigarette smoke which can induce oxidative stress and TGF-β have been shown to decrease HDAC2 activity.43 These findings suggest that oxidative stress and activation of PI3K-δ signaling may be important mechanisms for steroid resistance in bronchial asthma.
Immune mechanisms
Recent studies have demonstrated that Th17 cells and IL-17 levels appear to be increased in patients with severe asthma and linked to neutrophilic inflammation.44 Despite little information on a direct relationship with steroid resistance in airway disorders, murine Th17 cells seem to be steroid resistant.45 In addition, IL-17 increases the expression of GRβ in airway epithelial cells.33 Corticosteroids stimulate secretion of IL-10, an anti-inflammatory and immune-regulatory cytokine, and the decreased secretion of IL-10 in regulatory T cells has been reported in patients with steroid-resistant asthma.46 However, there is extremely scarce information on immune mechanisms associated with steroid resistance to date.
Endoplasmic reticulum stress in asthma
The ER is the major site of cells that is responsible for the synthesis, maturation, and trafficking of a wide range of proteins. Ca2+ homeostasis is also regulated in the ER.47 For appropriate protein production to occur, it is essential that physiochemical conditions within the ER lumen are adequate and molecular transport systems work well. When the ER is stressed by some conditions, such as increased demand in protein folding load in the ER lumen, cells evolve an adaptive response called UPR. For the normal molecular transport, the secretory pathway and the ER-associated degradation (ERAD) pathway should normally keep their intact systems with working.47 Any perturbations of these conditions, including failure of the ER's adaptive capacity, can reduce the ability of the ER to perform normal physiologic roles, causing ER stress.
In recent years, considerable evidence has demonstrated that ER stress is associated with pathogenetic mechanisms, especially inflammation within several disease, such as neurodegenerative disorders, metabolic disorders, cardiovascular diseases, malignancies, and respiratory disorders.3,48 Focusing on the respiratory system, several inhaled environmental triggers like cigarette smoke diesel exhaust, or allergens are known inducers of ER stress and cause dysregulation in ER homeostasis.49 Moreover, accumulating evidence has suggested the implications of prolonged ER stress and UPR in the development and progression of chronic lung diseases, including cystic fibrosis, α1-antitrypsin deficiency, idiopathic pulmonary fibrosis, pulmonary hypertension, chronic obstructive pulmonary disorders (COPD), and bronchial asthma.49 Our recent work has revealed that ER stress is important in the pathogenesis of bronchial asthma, especially steroid-resistant bronchial asthma at least in part through modulation of NF-κB.3 Interestingly, the results have indicated that the development of ER stress and activation of UPR in airway epithelial cells as well as inflammatory cells are critical in the induction and maintenance of severe asthma. In this review, we aim to highlight the role of ER stress in bronchial asthma, especially severe (steroid-resistant) asthma, concentrating on the related signaling pathways in airway epithelial cells.
Canonical UPR in the stress-conditioned ER
The canonical branches of UPR, which are conserved from yeast to humans, are mediated by activating 3 sensors that are localized in the ER membrane-inositol requiring enzyme (IRE) 1, activating transcription factor (ATF) 6, and double-stranded RNA-activated protein kinase-like ER kinase (PERK).50 According to the classic model of UPR activation, under basal conditions, theses 3 transmembrane proteins are bound by a chaperone, binding immunoglobulin protein (BiP)/glucose-regulated protein 78 (GRP78), which keep them in an inactive, monomeric state.51 Accumulation of misfolded proteins and increased protein cargo in the ER cause recruitment of BiP away from these UPR sensors. Activation of IRE1 and PERK leads to their dimerization and autophosphorylation, while ATF6 needs to translocate to the Golgi apparatus in order to become activated.47 These 3 proteins mitigate ER stress by reducing protein synthesis, facilitating protein degradation, and increasing production of chaperones that help proteins in the ER lumen to fold. As a result, ER stress resolves, and if it does not, then the cell is functionally compromised and may undergo apoptosis.52 Among them, IRE1α is the oldest and most conserved UPR branch in an evolutionary sense and possesses both activities of kinase and endoribonuclease.50 The kinase activity of IRE1α results in JNK activation,53 whereas the RNAse activity of IRE1α leads to the processing of mRNA encoding the transcription factor X-box-binding protein 1 (XBP1), with making XBP1 translated into an active (spliced) form.50 Spliced XBP1, alone or with ATF6α, mediates transcription of genes implicated in the production of chaperones, such as GRP78 and proteins involved in ER biogenesis, phospholipid synthesis, ERAD, and secretory pathways.50 In addition to the major pathway via XBP1 dealing with ER stress, recent studies have demonstrated that IRE1α also targets several other mRNAs for degradation through the regulated IRE1-dependent decay (RIDD) that is likely to be a new mechanism to explain the biological responses of ER stress.54
ATF6 belongs to a large family of cAMP response element binding (CREB)-like transcription factors that include ATF6α, ATF6β, CREB3L1 (also called OASIS), CREB3 (LUMAN), CREB3L2 (BBF2H7), CREB3L3 (CREBH), and CREB4.55 Activated ATF6 named as ATF6f, a cytosolic transcription factor, released from the Golgi apparatus in which ATF6 is cleaved by site 1 protease (S1P) and S2P enhances transcription of genes encoding ERAD components and XBP1.56 The family members of ATF6-like proteins also seem to be regulated by ER stress in a similar manner as ATF6 and appear to have distinct tissue distributions and tissue/cell type-specific functions, making a more delicate ER stress response possible in living things.52
Activation of the third arm of UPR, PERK results in phosphorylation of eukaryotic translational initiation factor 2α (eIF2α) which converts eIF2α into eIF2B, reduces global protein synthesis, and leads to a reduction in workload in the ER.57 In addition to a global reduction in protein synthesis, this branch of UPR is also linked to broad transcriptional regulation through activation of ATF4, nuclear erythroid 2-related factor 2 (Nrf2), and NF-κB that are master transcription factors with numerous functions, including regulation of the inflammatory response.58 ATF4 is produced through alternative translation and induces expression of genes involved in apoptosis (CHOP, C/EBP homologous protein), ER redox control (ERO1, ER oxidoreductin), the negative feedback release of eIF2α inhibition (Gadd34, growth arrest, and DNA damage-inducible protein), and glucose metabolism (fructose 1,6-bisphosphate; glucokinase; and phosphoenol pyruvate carboxykinase).59 Moreover, phosphorylation of eIF2α is mediated by not only PERK but also other kinases, such as protein kinase R (PKR), general control non-repressed (GCN2), and heme-regulated kinase (HKR). Although the role of other eIF2α kinases except PERK in ER stress remains unclear, studies to date have shown that PKR is activated under ER stress and influences UPR and/or related inflammatory signaling pathways.60 Various PERK-dependent or PERK-independent phosphorylation steps of eIF2α and the multiple targets of PERK suggest that UPR can be induced by various stimuli, such as oxidative stress and infectious condition, as well as ER stress due to improper protein synthesis and that inflammatory responses can be regulated by the PERK pathway through activation of a master transcription factor for inflammation like NF-κB. In fact, recent studies have reported that NF-κB activated by PERK regulates the production of inflammatory mediators, such as IL-6 and TNF-α.61
As mentioned above, UPR cannot always overcome ER stress and to restore the functional equilibrium of the ER. Under persistent and severe stressed conditions, UPR may induce cell death through apoptosis.62 The representative ER stress-associated apoptotic process is linked to the induction of CHOP which is activated via the PERK pathway. In addition, the JNK pathway and caspase-12 are activated, and proapoptotic BAX and BAK proteins are switched on by the IRE1α pathway.62,63 In addition, all the 3 canonical UPR pathways with differential activation can contribute to the induction of apoptotic responses when ER stress is excessive, persistent, or insufficiently resolved.47 However, more research is required to determine whether different arms of UPR are specialized to respond to particular conditions and different cellular environments by linking to distinct responses.
Although, the majority of studies on ER stress and UPR have focused on events evoked by the process of protein synthesis, many conditions as well as protein misfolding/accumulation can also induce ER stress and UPR, such as unbalanced ER calcium levels, metabolic abnormalities, hypoxia, oxidative stress, pathogens or allergens, pathogen-associated molecular patterns (PAMPs), and toxins. As for the respiratory system, several triggers of ER stress and UPR include cigarette smoke, airborne particulate matter (PM), bacterial infections, such as Pseudomonas aeruginosa and lipopolysaccharide (LPS), virus, house dust mite (HDM), and ovalbumin (OVA).3,49,64
ER stress and UPR in bronchial asthma
The underlying heterogeneity of bronchial asthma becomes a challenge to patients, physicians, and researchers who hope to develop treatments for severely and chronically ill patients. Thanks to many researchers who have provided a better understanding of endotypes and phenotypes of asthma, several novel targets have been suggested to be implicated in the pathogenesis of bronchial asthma, especially severe asthma, including steroid resistance.
A recent interesting study has proposed that a potential emerging molecular mechanism for bronchial asthma involves ER stress and UPR pathways3 which are linked to major inflammatory and stress signaling networks as well as production of reactive oxygen species (ROS) and NO.65,66 Moreover, that study has shown that the modulation of ER stress can overcome refractoriness of neutrophil-dominant asthma to steroids. Using an ER stress inhibitor, 4-phenylbutyric acid (PBA), airway hyperresponsiveness (AHR) and inflammation were significantly attenuated through inhibition of nuclear translocation of NF-κB in a mouse model of neutrophil-dominant asthma, while glucocorticoids did not have any significant effects on these asthmatic features and NF-κB activation. Furthermore, 4-PBA decreased TLR4 expression in various inflammatory cells, such as DCs, macrophages, and airway epithelial cells, whereas it increased the production of IL-10 in various airway inflammatory cells.
Based on genomic analysis, the most relevant candidate gene linking UPR to asthma is ORMDL3.67 The link between ORM (DL) proteins and UPR emerges from studies showing that orm1Δorm2Δ yeast displays a constitutive UPR.68 In addition, ORMDL3 modulates calcium signaling via association with the sarcoendoplasmic reticulum Ca2+ ATPase pump (SERCA), leading to reduced phospho-eIF2α.69 Moreover, inadequate SERCA expression has been shown to induce airway remodeling, a hallmark of asthma pathogenesis.70
In the lung, AGR2, a member of protein disulfide isomerase (PDI) family, is induced in asthmatic patients,71 and Agr2-/- mice display reduced levels of mucins MUC5AC and MUC5B during allergic airway inflammation.72 Another study has proposed that IRE-1β controls the levels of AGR2 and MUC5AC after exposure to allergens in an XBP1-dependent manner.73 In addition, IRE-1β coordinates the induction of genes involved in goblet cell differentiation and protein glycosylation, and inhibits Foxo2a forkhead box-family transcription factor 2a (Foxo2a) which suppresses the goblet cell phenotype.74 More interestingly, whereas Ire1β- and Agr2-deficient mice lack the mucin layer in the airway epithelium, additional parameters of asthma, including airway hyperreactivity or inflammatory cell recruitment, remain unaffected upon allergen challenge.72 Nogo is a protein family whose members are involved in shaping the tubular structure of the ER75 and is highly expressed in healthy lung tissue.76 However, in lung samples from asthmatic patients, Nogo expression is substantially decreased and in animal model showing the absence of Nogo, asthma symptoms are exaggerated.76 Moreover, when Nogo-B, an indirect target of ATF6 is reconstituted in epithelial cells it dampens excessive inflammation in this model,76 suggesting that ER integrity is essential in controlling the extent of Th2-medicated asthma.
As for the immunologic role of ER stress, among canonical UPR pathways XBP-1 was first identified as an essential transcription factor involved in the differentiation of plasma B cells.77 UPR has also been shown to be essential for the survival and development of DCs.78 Furthermore, ER stress seems to be a positive regulator of the immune response in patients with inflammatory pathologies.79 Actually, UPR activation has been reported to display a dramatically enhanced inflammatory response to TLR4 and TLR2 activation (i.e., induction of IL-23 and type I IFNs).80 In addition to the previous data on immune cells, our recent work has shown that ER stress-induced TLR4 expression occurs in various inflammatory cells infiltrating into lung tissue, airway epithelial cells, and several BAL cells, including macrophages. At the same time, primary cultured epithelial cells from OVA/LPS sensitized and OVA-challenged mice have shown that ER stress markers are overexpressed. These findings suggest that UPR pathways may couple innate to adaptive immunity in the lung, especially via TLR4 signaling in airway epithelial cells as well as other immune and inflammatory cells during allergic inflammation. Taken together, these data suggest that ER stress is of crucial relevance for establishing various forms of allergic asthma, including neutrophil-dominant and classic eosinophil-dominant asthma.
ER stress and steroid resistance in bronchial asthma
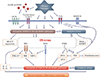
Accumulating evidence has indicated that ER stress and UPR are linked to major inflammatory and stress signaling networks, including the NF-κB pathway and oxidative stress. Interestingly, we have realized that the majority of these signaling networks are also involved in mechanisms associated with the development of steroid resistance. In this section, we discuss the role of ER stress and UPR pathways in the pathogenesis of severe asthma, particularly steroid resistance based on related inflammatory and stress signaling networks (Figure).
JNK/AP-1 signaling pathway and UPR
During ER stress, autophosphorylation of IRE1α triggers its binding to TNF receptor-associated factor 2 (TRAF2), and the IRE1α-TRAF2 complex can interact with JNK, which activates transcription factor AP-1, resulting in the expression of inflammatory genes.53 As mentioned above, AP-1 is considered the most important transcriptional factor associated with steroid resistance of asthma because AP-1 physically interacts with GR, thereby preventing its binding to GREs and other transcriptional factors.35 In addition, levels of AP-1, phosphorylated JNK, and c-Fos are elevated in inflammatory cells from glucocorticoid-resistant asthmatics.35 Our previous study showed that phosphorylation of JNK and ERK as well as p38 MAPK was significantly increased in lung tissues of OVA-inhaled mice compared with control mice.81 When the potent antioxidant was administered to the mouse model of asthma, JNK activation was not affected, whereas the p38 MAPK pathway was significantly inhibited and asthmatic features were attenuated, indicating that the JNK pathway is activated in allergic airway inflammation; however, the symptom controller does not suppress all the activated inflammatory signaling pathways. A product of UPR, XBP-1, which can be produced by IRE1α activation-JNK phosphorylation was more expressed in the lung tissues of OVA-LPS-sensitized and OVA-challenged mice showing neutrophil-dominant inflammation than in those control mice.3 Increased XBP-1 expression was significantly reduced by the administration of 4-PBA, while administration of dexamethasone showed no effects on the expression of ER stress marker, GRP78 and CHOP. Taken together, although further studies are required to determine whether this UPR-JNK-AP-1 signaling pathway is linked to the development of the severe asthmatic phenotype through steroid resistance, these findings suggest the potential of ER stress as a promising target for the control of the JNK-AP-1 signaling pathway in airway inflammation.
NF-κB signaling pathway and UPR
Three canonical UPR pathways appear to be tightly interrelated with the NF-κB signaling pathway. In fact, our recent studies on pulmonary disorders have revealed that ER stress activates the NF-κB signaling pathway which explosively evokes proinflammatory gene expression and cytokine production in the lung.3,64 In our studies, both animal models for LPS-induced acute lung injury and OVA-LPS induced neutrophil-dominant asthma showed marked increases in the nuclear translocation of NF-κB in lung tissues, and the increases were substantially blocked by administration of 4-PBA with attenuation of pathophysiological abnormal features, such as inflammation or plasma exudation. In addition, ATF6 is linked to the NF-κB-IκB kinase (IKK) signaling pathway.82 Moreover, in response to ER stress, PERK-eIF2α-mediated suppression of IκB translation can induce expression of inflammatory cytokines, allowing the excess of free NF-κB to enter the nucleus. IRE1α activation also recruits of IKK and promotes NF-κB-mediated inflammation.61 Intriguingly, in the mouse model of steroid-resistant asthma, increases in ER stress markers, GRP78 and CHOP, as well as a UPR branch, ATF6α, were dramatically reduced by treatment with 4-PBA, while dexamethasone showed no effects on their expression in lung tissues.3 In particular, in LPS-stimulated airway epithelial cells, we found that increased NF-κB nuclear translocation was inhibited by pretreatment with 4-PBA [unpublished data]. Given the relationship between the ER stress-UPR pathway and the NF-κB signaling pathway, these findings indicate that, in certain conditions, steroid refractoriness to control NF-κB activation can be overcome by the blockade of ER stress or the UPR pathway in airway epithelial cells as well as inflammatory cells.
PI3K signaling and UPR
Phosphatidylinositol is one of the phospholipids synthesized in the ER and in highly dynamic ER-derived compartments, and phosphorylated phosphatidylinositol, i.e., PIPs are the regulators of vesicular transport and secretary pathway of the ER.83 Therefore, it can be hypothesized that, on ER stress induced by any stimuli, the PI3K signaling pathway can be, to some degree, affected by improper production of PIPs in the ER and its transport system, although it is controversial whether abnormal responses lead to activation or suppression of PI3K signaling. A previous study has revealed that the PI3K/Akt signaling pathway is required for the accumulation of GRP78 whose expression is primarily regulated by ATF6 and XBP-1 responses.84 Other interesting studies have demonstrated that p85, a regulatory subunit of PI3K, regulates XBP-1, in which the interaction between p85 and spliced XBP-1 leads to spliced XBP-1's nuclear translocation and possibly stabilization.85 Actually, overexpression of p85 in a mouse model for obesity and diabetes restores spliced XBP-1 nuclear translocation and UPR expression in response to feeding stimuli.85 Specifically, inhibition of PI3K-δ pathway serves as a critical target for treating glucocorticoid-resistant severe asthma as well as non-severe asthma.1 Interestingly, we found that a mouse model of mold-induced asthma showed exaggerated ER stress and UPR pathway activation in the lung, which were significantly blocked by treatment with a PI3K-δ inhibitor, IC87114 [unpublished data], suggesting that PI3K-δ is linked to the modulation of ER stress in fungus-related allergic lung inflammation. In addition, LPS-stimulated airway epithelial cells showed increased PI3K-δ or -γ expression, including the p85 regulatory and p110 catalytic subunits [unpublished data]. Altogether, the interplay between ER stress and PI3K-δ can be a novel target for the control of bronchial asthma, especially severe steroid-resistant asthma.
PKR phosphorylation and UPR
PKR is well characterized as an essential component of the innate antiviral response. In addition, a recent study has indicated that PKR is implicated in TLR signal transduction in response to bacterial cell wall components.86 As described above, PKR can also phosphorylate eIF2α which is one of the major targets of PERK, a UPR branch, leading to activation of various inflammatory responses. Exposure to pathogens/allergens containing TLR4 ligands results in XBP1 activation in alveolar macrophages, DCs, and airway epithelium.73,80 In addition, viruses expressing glycoproteins activate IRE1α and PERK87; lung epithelial cells infected with some viruses up-regulate GRP78 and initiate apoptosis in a caspase-12-dependent manner.88 Activation of the TLR4/MyD88 pathway inhibits GR nuclear translocation in murine pulmonary macrophages, resulting in steroid resistance,27 and TLR2/TLR4 signaling induces PKR phosphorylation switching in their downstream intracellular signaling related to ER stress and inflammatory responses.86 These findings provide a new interesting mechanism for steroid resistance in asthma and/or pulmonary disorders.
Metabolic abnormalities and inflammation: Metaflammasome, IGF-I/IGFBP-3, PPARγ, and UPR
The concept that acute/chronic inflammation and immune responses are mechanisms underlying the development of various metabolic disorders such as a fatty liver, obesity, diabetes mellitus, and cardiovascular disorders, has been accepted as valuable information.89 In addition to pathogen-associated stimuli, PKR is markedly induced by lipids, especially in the obese state and plays a critical role in the activation of JNK and inflammatory responses.52,60 PKR is a core component of the metaflammasome (metabolic inflammasome) and interacts directly with several inflammatory kinases, such as IKK and JNK, insulin receptor signaling components, such as IRS1, and the translational machinery via eIF2α related to metabolism and inflammation.52 Formation of metaflammasome and its activation by nutrients and ER stress may explain the functional overlap between metabolic and inflammatory signaling.
Concentrating on bronchial asthma, obesity-associated asthma is one of the endotypes in relation to severe asthma and shows female predominance, very late onset, and poor therapeutic responses to steroids.2 Given that insulin metabolism can affect glucose transport, lipid accumulation, and the development of obesity, it is very interesting that the insulin-like growth factor (IGF)-1/insulin-like growth factor-binding protein (IGFBP) system contribute to the pathogenesis of bronchial asthma. In fact, IGF-I has been reported to play an important role, especially in subepithelial fibrosis, airway inflammation, AHR, and airway smooth hyperplasia, indicating that regulation of the IGF-I signaling pathway may have the therapeutic potential for asthma.90 Moreover, recent studies have also shown that IGFBP-3 plays a critical role in asthmatic inflammation through an IGF-I-dependent and/or IGF-I-independent mechanism.91 When IGF-I binds to the IGF-I receptor, the kinase domains of the receptor are phosphorylated.92 This process induces phosphorylation of binding sites for docking proteins, such as IR substrates 1 to 4 (IRS1 to IRS4) which is one of the signaling components of metaflamasome, and IRS1 is known to stimulate various signaling pathways, such as MAPK and PI3K pathways.93 Based on the relationship between ER stress and metaflammasome, these findings suggest that the IGF/IGFBP system is one of the integrated networks to ER stress and provides an exciting target to achieve better management for bronchial asthma, especially the obesity-associated asthmatic endotype.
In view of metabolic inflammation, we can recall another signaling pathway implicated in the pathogenesis of bronchial asthma. The peroxisome proliferator-activated receptor (PPAR)γ is a family member of nuclear receptor ligand-activated transcription factors which participate in the maintenance of lipid and glucose homeostasis and control of cell growth and differentiation.94 At the same time, the PPARγ and its ligands are known to have anti-inflammatory properties, such as inhibition of cytokine signaling, transcription factor activation, and inflammatory gene expression.95 Furthermore, several previous studies have demonstrated that treatment with PPARγ agonists, such as pioglitazone and rosiglitazone, has positive therapeutic effects in airway inflammation, AHR, and airway remodeling in an animal model of asthma.96 Although the effects of PPARγ ligands on ER stress are controversial and different depending on cell types and tissues, the PPARγ has been reported to exhibit its anti-inflammatory property through modulation of ER stress.97 Taken together, in the field of metabolic abnormality-associated bronchial asthma, especially obesity, efforts to investigate the interaction between the PPARγ and ER stress in airway inflammation may provide us a novel therapeutic strategy for each endotype of bronchial asthma.
Immune response and UPR
Recent studies have reported that pathogenetic mechanisms of steroid-resistant asthma is evolved in the expression/action of IL-17 and IL-10 in the lung.45,46 Supporting this contention, our recent work has shown that the expression of IL-17 and IL-10 is significantly increased in lung tissues and BAL fluids of mice with steroid-resistant neutrophil dominant asthma.3 Administration of 4-PBA, an ER stress inhibitor, dramatically reduces the increased expression of IL-17, whereas it further enhances the increase in IL-10 levels, resulting in attenuation of asthmatic features. Moreover, additional experiments using LPS-stimulated airway epithelial cells revealed a positive feedback interaction between IL-17 and ER stress [unpublished data]. Taken together, it is expected that the modulation of ER stress and UPR can be a powerful tool to surpass the limit of steroids in treatment for bronchial asthma.
Oxidative stress and UPR
Nowadays, both oxidative and nitrative stresses have been accepted as crucial for the development of bronchial asthma. In addition, they also contribute to the development of steroid resistance in chronic airway inflammatory disorders, such as bronchial asthma and COPD through several mechanisms, including enhancement of proinflammatory transcriptional factors, such as PI3K-δ, NF-κB, AP-1, and HIF-1α,42,81 and a reduction in HDAC2 expression/activity.41
Meanwhile, oxidative stress is closely linked to ER stress, disrupts protein folding, and then activates UPR pathways, including PERK and vice versa.65,98 In fact, growing evidence suggests that ER stress, oxidative stress, and inflammatory responses are cross-linked and that limiting of either one will affect the others via messengers, such as ROS, calcium, and NO.98 In interactive signal transduction processes, functions of the ER and mitochondria are closely linked. These 2 organelles build up a dynamic network in which they generate calcium signals and ROS to stimulate ER stress, oxidative stress, and inflammation.99 In mold-induced asthmatic inflammation of mice, we found that ER and oxidative stresses were increased and that when oxidative stress was reduced by treatment with a mitochondrial ROS inhibitor, ER stress was also substantially decreased. In the same study, treatment with 4-PBA also effectively reduced the generation of mitochondrial ROS [unpublished data]. These findings may establish the mechanism of a cross-talk between ER and oxidative stresses, providing a potential target for controlling bronchial asthma, even with steroid resistance.
Therapeutic potential of targeting er stress and perspectives in severe asthma
The last decade has witnessed an explosion in the elucidation of causative mechanisms implicated in bronchial asthma, especially severe or steroid-resistant asthma; however, treatment of asthmatic patients is still challenging. One of the reasons stems from the absence of adequate animal models representing real-life of allergic patients and showing steroid resistance. In addition, many new treatments specifically target a single mediator or receptor and are unlikely to have a major clinical effect, although they may be effective in specific asthma phenotypes. Drugs with more widespread effects, such as kinase inhibitors, may be more effective but have a greater risk of side effects.100 Therefore, new treatments should be contemplated to target multiple underlying allergic/immune processes and minimize adverse effects on other systems. Recently, accumulating findings suggest that regulation of ER stress is a prospective molecular therapeutic target for various pulmonary disorders, including bronchial asthma. A more encouraging finding is that inhibition of ER stress overcomes the failure of steroids to attenuate the severe asthmatic features of mice. Furthermore, as described above, therapeutic approaches to ER stress can concomitantly regulate multiple integrated signaling networks known as well-known pathogenetic mechanisms for inflammatory disorders. Despite success in mice, to date, there is the shortage of information on molecular mechanisms explaining these effects of the ER stress control, and there are also no clinical trials to evaluate the therapeutic effects of pharmacologic agents targeting ER stress in humans. Finally, since almost all the data are derived from animal studies or cell culture ex-vivo, well-organized and large-scaled clinical trials are essential in verifying the strengths and superiority of agents targeting ER stress for better management of bronchial asthma, especially severe asthma.
In conclusion, regulation of ER stress is a potentially exciting target for developing agents to achieve better management of bronchial asthma, especially severe asthma in which steroids and other current agents are less effective.
 XML Download
XML Download