

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Lysophosphatidylcholine (LPC) is a lipid mediator with pro-inflammatory and pro-atherogenic activities. It is believed to be a critical factor underlying cardiovascular diseases,1,2 and several pathological conditions are associated with elevated LPC levels in the circulation.3-5 LPC has a broad-spectrum of pro-inflammatory activities, including promotion of cell growth,6 migration,7,8 secretion of chemokines and cytokines,9,10 generation of reactive oxygen species,11 and upregulation of adhesion molecules such as ICAM-1, VCAM-1, and selectins.12
Evidence implicates involvement of LPC in inflammatory injury of lungs, such as acute lung injury and chronic diseases such as asthma. In lungs from late stage adult respiratory distress syndrome,13 lungs challenged with antigen14 or instilled with lipopolysaccharide,15 elevated amounts of LPC are reported in the broncho-alveolar lavage fluids (BALF). Furthermore, the administration of exogenous LPC into lungs of animal models promotes eosinophil infiltration,16 increases airway resistance,16,17 and increases airway epithelial permeability.18,19 These findings suggest that elevated levels of LPC in the extracellular lung fluids can induce inflammatory injury on the airway and alveolar cell populations. To date, the role of LPC in lung diseases is incomplete.
Asthma is a chronic inflammatory disease of the airways characterized by epithelial injury, leukocytic infiltration, airway hyper-reactivity, and airway wall remodeling. We postulate that the chronic inflammation is attributed, at least in part, to elevated amounts of LPC in the lungs. In lungs of asthmatic subjects14,20-22 and in animal models of asthma,23,24 the LPC-generating enzyme, phospholipase A2 (PLA2) is significantly increased. PLA2 hydrolyzes membrane phosphatidylcholines and oxidized phosphatidylcholines at the sn-2 position, generating LPC following release of the free fatty acid.25 However, it is not known whether the inherent LPC content in lungs is increased in asthmatics. To begin to investigate the involvement of LPC in asthma, this study determined LPC levels in the BALF from asthmatic subjects.
MATERIALS AND METHODS
BALF collection and preparation
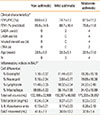
Eight non-asthmatic controls and seven asthmatic subjects were recruited for bronchoscopy and BALF collection under an approved protocol by Institutional Review Board. The asthmatics were further grouped as mild or moderate based on spirometric measures of forced expiratory volume in 1 sec (FEV1) and/or clinical features (Table 1). The BALF was filtered to remove mucus and visible cellular debris, and an aliquot used for cell count determination. The remaining BALF was centrifuged in which the supernatant was stored at -80℃ for later analysis of LPC content, protein concentration, and PLA2 activity; whereas the cell pellet for qRT-PCR of PLA2.
Quantification of LPC species
The supernatant fraction of the BALF was extracted for quantification of the 3 most common and biologically-active LPC species (LPC16:0, LPC18:0, and LPC18:1) by high performance liquid chromatography-tandem mass spectrometry (LC-MS-MS). The samples were spiked with LPC19:0 (140 ng/mL) as an internal standard. The system consisted of a Thermo TSQ Quantum triple quadrupole mass spectrometer (San Jose, CA) equipped with a Waters 2695 HPLC system (Milford, MA) and a Waters XTerra C18 column, and has a detection limit for LPC of 10 pg. The limit of detection of LPC was 10 pg and the limit of quantitation was 25 pg. The standard curves for the LPCs over the concentration range of 2.5-500 ng/mL were linear with a coefficient of determination (R2) >0.995.
Microplate assays
PLA2 activity was measured from the supernatant fraction of BALF using a fluorescent phosphatidylcholine substrate analog (Red/Green BODIPY PC-A2). Cleavage of the substrate analog by PLA2 results in increased fluorescence detected at 515 nm. The chemokine, IL-8, was measured as a prototypic inflammatory marker in the BALF using an enzyme-linked immunoabsorbent assay (ELISA) kit (BioLegend). Absorbance was read at 450 nm.
Quantification of PLA2 mRNA
The cell fraction of the BALF was evaluated by qRT-PCR as a potential source of secreted PLA2. Total RNA was isolated using Trizol according to manufacturer's instructions, quantified by absorbance at 260 nm (NanoDrop DNA/RNA/protein spectrophotometer), and cDNA synthesized using the high capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA). The cDNA was amplified with SYBR® Green PCR master mix kit (Applied Biosystems) using the BIORAD MyiQ single color real-time PCR detection system (Hercules, CA ) in a 96-well format. Primers were designed to recognize major types of human PLA2 and the internal control gene, glyceraldehyde-3-phosphate dehydrogenase as follows: sPLA2-IIa (NM_000300), sPLA2-V (NM_000929), sPLA2-X (NM_003561), sPLA2-XII (NM_030821), cPLA2-IVa (NM_024420), iPLA2-VI (NM_003560), GAPDH (NM_002046).
RESULTS
Table 1 summarizes the subjects' clinical characteristics, of which the lung function data (FEV1) were used to categorize subjects into non-asthmatic control, mild- or moderate-asthmatics according to guidelines from the Global Initiative for Asthma.26 As expected, the BALF in the mild- and moderate-asthmatics presented with several indices of inflammation (Table 1). A cell differential analysis of BALF from mild-asthmatics indicated higher percentages of eosinophils and lymphocytes relative to non-asthmatic controls. These proportions of leukocytes further increased in the moderate-asthmatics, which now included neutrophils. An upward trend was observed in the total cell count and protein content in the BALF of mild- and moderate-asthmatics. We also found a progressive increase of IL-8 content in the non-asthmatics to the mild- and moderate-asthmatics. In summary, the BALF of asthmatics contain several markers of inflammation that progress with degree of asthma severity.
We found that non-asthmatics have basal levels of all 3 LPC species at a range of 180-250 ng/mL of BALF (Fig. 1). The saturated species LPC16:0 and LPC18:0 were significantly increased in asthmatic subjects with FEV1 characteristic of moderate asthmatics (Table 1) when compared with the non-asthmatic controls (Fig. 1). Asthmatic subjects with less severe FEV1 representing the mild-asthmatics showed no increases of the LPC species relative to controls. The findings indicate that the lung mucosal surface of moderate-asthmatics is likely to be chronically exposed to ~1.3 to 1.9-fold higher amounts of LPC16:0 and LPC18:0, respectively, than in non-asthmatics.
We next determined whether the increased LPC content in lungs of moderate-asthmatics could be attributed to secreted PLA2, hydrolyzing phosphatidylcholines of cell membranes or surfactant. Measures of the PLA2 activity of BALF indicated that moderate-asthmatics, but not mild-asthmatics, show a 3-fold increase over non-asthmatics (Fig. 2A). The infiltrated lung leukocytes as well as the airway epithelium are the likely cell populations that secrete PLA2 into the lung luminal fluids. Quantitative RT-PCR evaluation of the cell fraction of the BALF indicated that subtype secretory sPLA2-X mRNA was upregulated in moderate-asthmatics, but there were no changes in other subtypes (sPLA2-XII, cPLA2-IVa and iPLA2-VI) (Fig. 2B). These findings suggest that sPLA2-X is likely a key subtype in the generation of LPC in asthmatic subjects.
DISCUSSION
The central new finding is that inherent levels of LPC16:0 and LPC18:0, but not LPC18:1, were significantly elevated in lung lining fluids of subjects with lung function impairment characteristic of moderate asthma. These corresponded to 1.3- and 2-fold respective increases in LPC16:0 and LPC18:0 over non-asthmatic controls. Interestingly, in atopic asthmatic subjects challenged with antigen, LPC16:0 is increased in BALF when compared to saline challenge;14 however, the LPC level in non-asthmatic control was not reported to ascertain the effects in the absence of antigen challenge in asthmatics. Reports indicate that among acyl-lysophospholipid species, LPC16:0 and LPC18:0 exert the greatest pro-inflammatory activities, such as eosinophil adhesion,27 neutrophil priming,28 and cytokine secretion.9,10 Altogether, the results implicate a pathogenic role of LPC in the chronic inflammatory injury of the asthmatic airway.
We can only speculate on the inflammatory potential of an elevated LPC content in lung lining fluids of moderate-asthmatics, since precise in vivo LPC concentrations are unknown and its bioactivity is dependent on the free form (not bound to albumin or other proteins).1 In in vitro culture studies, LPC concentrations of 1-100 µM are effective in inducing a wide range of inflammatory activities, such as cell proliferation,6 migration,7,8 increases in endothelial permeability,29 leukocyte adhesion,27,30 neutrophil priming,28 and cytokine secretion.9,10 The calculation of molar units of LPC from our data indicated 70 µM of LPC16:0 and 140 µM of LPC18:0 from moderate asthmatics, which are values comparable to those used in in vitro studies. This finding suggests that the increased LPC content in lungs of moderate-asthmatics could be sufficient in promotion of airway inflammatory injury.
The results indicate that the increased LPC content in lungs of moderate-asthmatics was accompanied by significant increases in the PLA2 activity in the lung lining fluids, providing evidence that increased generation of LPC was likely attributed to secreted PLA2. Moreover, the PLA2-induced increase in LPC is expected to be accompanied by concomitant equimolar amounts of free fatty acids, which can be arachidonic acid depending on the substrate phosphatidylcholine species. The well-recognized pathway of arachidonic acid metabolism by cyclooxygenases and lipoxygenases will, in turn, produce respective prostaglandins and leukotrienes, which are believed to be important lipid mediators in asthma. Thus, increases in LPC together with increases in arachidonic acid metabolites suggests that the inflammatory potential of PLA2 is likely significantly greater than recognized previously.
Furthermore, the increased PLA2 activity was attributed to, at least in part, to upregulated sPLA2-X mRNA in the leukocytic infiltrate, a finding consistent with the report of overexpression of sPLA2-X in airway epithelial cells and bronchial macrophages of human asthmatic subjects.20 Although our protocol did not allow us to identify the cell type(s) responsible for the upregulated sPLA2-X, analysis of the cell differential indicated that in moderate-asthmatics, the infiltrate population shifted towards increasing eosinophils, neutrophils, and lymphocytes (Table 1).
In summary, the lung lining fluids in moderate-asthmatics showed an inherently elevated content of LPC16:0 and LPC18:0. Such increased LPC is a potential injurious mediator in the initiation and/or progression of airway inflammatory injury in asthma.


 XML Download
XML Download