

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The mast cell has long been known to be a key effector cell type in IgE-mediated immediate hypersensitivity and allergic disorders,1,2 although recent studies have added their novel roles in host defense against bacteria and bee and snake venoms, as well as immunoregulatory roles.3 Mast cells are activated by cross-linking of IgE-bound high-affinity IgE receptors (FcεRI) with multivalent antigen.4 Activated mast cells secrete preformed proinflammatory mediators, such as histamine, serotonin, nucleotides, proteases, and tumor necrosis factor (TNF)-α, and synthesize and secrete various cytokines, chemokines, and lipid mediators, such as leukotrienes and prostaglandins. The past two decades showed tremendous progress in our understanding of what happens inside the mast cells whose activation is triggered by IgE plus specific antigen.5-7 Thus, many scientists, particularly those interested in FcεRI signal transduction research from outside the field, seem to be inclined to think that we have a very good understanding of how mast cells are activated through the allergen-IgE-FcεRI axis.
In contrast to the long-held view of the requirement of multivalent antigen (or anti-IgE antibody) to cross-link IgE-bound FcεRI, we and others have demonstrated that monomeric IgE binding to FcεRI in the absence of specific antigen elicits numerous biological activities in mast cells: upregulated cell surface expression of FcεRI,8-10 survival,11,12 increased histamine content,13 histamine release, leukotriene release, receptor internalization, DNA synthesis,14 increased responses to compound 48/80 and substance P,15 increased filamentous actin content,16 membrane ruffling,17 adhesion to fibronectin,18 and migration.19 Similar to cells stimulated with IgE plus antigen, FcεRI cross-linking appears to be essential for mast cell activation induced by monomeric IgE.14
HETEROGENEITY OF IgE MOLECULES
Two papers published in 2001 showed the same effects of monomeric IgE, i.e., survival promotion, as well as disparate results. Kalesnikoff et al.12 showed that monomeric IgE could induce cytokine secretion and the corresponding signaling data, whereas Asai et al.11 reported the absence of cytokine production. A follow-up study revealed that this difference stems from the difference in the IgE molecules used.14 Importantly, IgEs display a wide range of heterogeneity in their ability to induce the production and secretion of IL-6 and TNF-α and other activation events in mouse mast cells. At one end of the spectrum, highly cytokinergic (HC) IgEs can induce not only strong survival promotion but also all other activation events listed in the preceding paragraph, whereas, at the other end of the spectrum, poorly cytokinergic (PC) IgEs can induce only weak survival enhancement.14 More extensive receptor aggregation can be induced by HC IgEs than by PC IgEs. Polyclonal IgE from mice and humans suffering from atopic dermatitis (AD) can enhance survival and cytokine production in mast cell cultures, indicating that monomeric IgE effects are operative in polyclonal situations as well.20
The difference between HC and PC IgEs appeared to be similar to the IgE+/IgE- dichotomy with regard to the priming ability of IgE in response to the relatively unknown cytokine-like protein histamine-releasing factor (HRF). HRF is an evolutionally conserved protein with a molecular mass of ~26 kDa.21 Prior to the purification and cloning of HRF in 1995, HRF-like activities had been reported from a variety of cellular sources including alveolar macrophages, platelets, vascular endothelial cells, B and T lymphocytes, mononuclear cell cultures, the U937 monocyte/macrophage-like cell line, and the RPMI 8866 B cell line.22 HRF-like activity was also found in nasal lavages, skin blister fluids, and bronchoalveolar lavage fluids during the late phase of allergic reactions.23-25 Without a signal sequence, the secretion of HRF is insensitive to brefeldin A or monensin, but it can be enhanced by TSAP6, a p53-inducible 5-6 transmembrane protein.26 HRF can be found in exosomes, suggesting that it is secreted through a nonclassical exosome pathway.26 Because human HRF can stimulate histamine release and cytokine (IL-4 and IL-13) production from IgE-sensitized (but not IgE-depleted) basophils and mast cells,21,27,28 it is considered an IgE-dependent cytokine. MacDonald et al. revealed functional heterogeneity among human IgE molecules: IgE from HRF-responder (HRF-R) basophils derived from ~50% of atopic patients was termed IgE+, and IgE from nonresponders (HRF-NR) was termed IgE-.29 However, heterogeneity in the carbohydrate portion of IgE molecules failed to distinguish between IgE+ and IgE-.30 Interestingly, HRF responses in human basophils were shown to be negatively correlated with SHIP (SH2 domain-containing phosphatidylinositol 5'-phosphatase), but not Syk (spleen tyrosine kinase), levels,31 explaining some HRF-R subjects. Despite these efforts, the exact molecular basis of the IgE+/IgE- dichotomy remained an enigma for a number of years.
HRF BINDS A SUBSET OF IgE AND IgG MOLECULES
The most obvious interpretation of the two similar phenomena of IgE heterogeneity leads to the idea that some, but not all, IgE molecules interact with HRF. However, Wantke et al. thought that HRF does not bind to IgE, because they previously observed that HRF primed anti-IgE-antibody-induced histamine release from all basophils, irrespective of the type of IgE on the cell surface.32 So, they performed experiments using a rat basophilic leukemia cell line (RBL-SX38), which was engineered to express a functional human FcεRI. They found that neither human nor mouse recombinant HRF caused histamine release in RBL-SX38 cells sensitized with IgE+. Additionally, priming the transfected RBL-SX38 cells or the parental cell line, RBL-2H3 cells, with human or mouse recombinant HRF did not increase anti-IgE-induced histamine release. Based on these results, the authors concluded that HRF does not bind to either IgE+ or IgE-.
Because we later (in 2003) found the heterogeneity of IgEs in their monomeric effects,14 we decided to pursue the most obvious possibility, namely IgE-HRF binding. However, being concerned this time that only a small proportion of IgEs might interact with HRF, we tested a relatively large panel of IgE mAbs. Simple ELISA experiments using GST-mHRF (glutathione S-transferase [GST] fused to mouse HRF) as a capturing agent, our experiments showed that six of 19 IgE mAbs tested bound GST-mHRF but not GST. Some of the HRF-reactive IgEs were also used to confirm HRF binding by two other methods. Similar proportions of IgG mAbs also bound GST-mHRF. Binding properties were not determined by antigen specificity or IgG isotype. Affinity measurement using the quartz crystal microbalance method33 indicated that these interactions were of low affinity (KD=~10-6 M).
DEVELOPMENT OF HRF INHIBITORS AND THEIR USE TO INHIBIT ALLERGIC INFLAMMATION
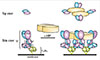
HRF-binding sites were mapped to Fab, not Fc, portions of IgE and IgG molecules. The HRF-binding sites seemed to be close to or to overlap at least partially with antigen-binding sites. On the other hand, IgE- (and IgG-) binding sites were mapped to the N-terminal 19 residues (N19) and the H3 (residues 107-135) portion of mHRF. Importantly, both GST-N19 and GST-H3 could inhibit HRF-IgE (or IgG) interactions in ELISA (Fig. 1). We also confirmed previous data showing that HRF can be present as a disulphide-bonded dimer,34 suggesting that HRF should be able to induce cross-linking of HRF-reactive IgE-bound FcεRI (Fig. 2), which was supported by mast cell activation induced by IgE plus HRF. This activation could be inhibited by GST-N19 or GST-H3. These results suggested that GST-N19 and GST-H3 may be useful reagents to probe the function of HRF in in vivo models of allergic diseases. However, they are required to not affect HRF's intracellular functions (see Box) because those functions are essential for fundamental cellular properties such as proliferation and survival. Fortunately, neither GST-N19 nor GST-H3 affected the growth or survival of various cultured cells; this was shown to be due to their failure to enter the cell.
Using these HRF inhibitors, we could show that passive cutaneous anaphylaxis (PCA) induced by intradermal antigen challenge in mice primed with HRF-reactive IgE can be substantially ameliorated by GST-N19 or GST-H3. Combined pretreatment with GST-N19 and GST-H3 largely inhibited PCA reactions. We also demonstrated that pretreatment with GST-N19 before allergen (i.e., ovalbumin [OVA]) challenge strongly inhibited OVA-induced mast cell-dependent airway inflammation, as revealed by reduced eosinophilia in the airway, reduced immune cell infiltration in the lungs, reduced IL-5 and IL-13 in the lungs, and reduced airway hyperresponsiveness. Synthetic N19 peptide also showed similar efficacy to inhibit airway inflammation. However, GST-N19 was ineffective in inhibiting T cell-dependent (but mast cell-independent) airway inflammation induced by OVA challenge in OVA+alum-sensitized mice. A partially IgE-dependent model induced by intranasal administration of Aspergillus fumigatus extract was partially inhibited by GST-N19. Therefore, these results suggest that mast cells are target cells for HRF to promote airway and skin inflammation induced by IgE.
MECHANISM OF HRF-MEDIATED INFLAMMATION
The efficacy of HRF inhibitors in the suppression of PCA induced by HRF-reactive IgE (anti-TNP IgE mAb C38-2) and antigen (TNP-BSA) can be understood as follows: IgE-antigen interactions may be inhibited by HRF inhibitors because the interaction of HRF-reactive IgE with HRF was at least partially inhibited by monovalent TNP hapten. However, it is much more difficult to understand how HRF affects airway inflammation because airway inflammation involves a complex interplay of various types of cells and many soluble and insoluble factors. Therefore, we pursued the development of a simpler protocol to study HRF-mediated inflammation. Intranasal administration of recombinant mHRF in naïve mice has been shown to induce weak lung inflammation. Similar to transgenic mice overexpressing HRF in a Clara cell-specific manner,35 this lung inflammation consists mainly of macrophages. Using various mutant mice, we could show that this inflammation required B cells (source of immunoglobulins [Igs]) and mast cells (target cells), as well as FcRγ. FcRγ is shared by multiple Fc receptors including FcεRI, FcγRI, FcγRIIIA, and FcγRIV.36,37 Among the Igs and Fc receptors, IgE and FcεRI were the predominant contributors to the HRF effects, as HRF-induced lung inflammation was abrogated in naïve FcεRI-/- mice. These results were consistent with the idea that IgE (and IgG) was the long-sought receptor for HRF. Global gene expression analysis was instrumental in further strengthening this idea. The expression of 196 genes was up- or down-regulated by over three fold by HRF in the lungs of naïve WT mice. Upregulated genes encode Th1-, Th2-, and Th17-associated cytokines and various chemokines, potentially accounting for the recruitment of monocytes/macrophages, neutrophils, eosinophils, and other immune cells. In contrast, only a small fraction of these genes (39 of 196 genes) fluctuated by more than three fold in FcεRIα-/- mice; furthermore, fewer (11 of 196 genes) genes were changed in FcRγ-/- mice. These results suggest that HRF executes its action largely, if not exclusively, by engaging IgE- and IgG-bound Fc receptors and promotes airway inflammation. Collectively, these findings solved three major HRF-related problems. First, the HRF receptor was identified. Second, the key role of HRF in allergic disease models was demonstrated. Third, the IgE+/IgE- dichotomy is now understood as the dependency of IgEs on their reactivity with HRF. What about other HRF functions? By definition, the functions of HRF should be limited to those of the extracellularly secreted protein, but not those of the intracellular protein TCTP (see Box).
Box. Intracellular functions of HRF (TCTP) versus extracellular functions of HRF
Despite the name "translationally controlled tumor protein", TCTP expression is subject to both transcriptional and translational control. TCTP within a cell plays a critical role in the fundamental processes of cell-cycle progression, proliferation, survival, and malignant transformation. TCTP is a Ca2+-binding protein.60-63 It is involved in the elongation step of protein synthesis by interacting with both eEF1A (a small GTPase) and eEF1Bβ (a guanine nucleotide exchange factor).64-66 Drosophila TCTP acts as the guanine nucleotide-exchange factor for Rheb (Ras homologue enriched in brain), a Ras superfamily GTPase that regulates the TSC1-mTOR pathway.67 TCTP interacts with Mcl-168,69 and Bcl-xL,70 anti-apoptotic members of the Bcl-2 family. Recent studies identified mutual antagonism between p53 tumor suppressor and TCTP, with TCTP promoting p53 degradation via MDM2-mediated ubiquitination of p53 and with p53 directly repressing TCTP transcription.71,72 Another conserved property of TCTP is its interaction with microtubules and mitochondria.73 Readers interested in this topic are referred to Bommer.74
TCTP and HRF are the same protein. The extracellular functions of the protein are the focus of this review. As discussed in the text, they include the activation of mast cells and basophils whose FcεRI molecules are occupied with HRF-reactive IgE.
CAN WE EXPALIN ALL KNOWN PROPERTIES OF HRF?
Much of the 'HRF enigma' has been solved. However, HRF is also known as a B cell growth factor.38 HRF can stimulate IL-8 secretion from GM-CSF-primed eosinophils.39 HRF can inhibit PMA+A23187-stimulated cytokine gene expression in T cells.40 More recently, HRF has been shown to stimulate bronchial epithelial cells to produce IL-8 and GM-CSF.41 It is tempting to speculate that these HRF functions could occur through HRF's interactions with B cell antigen receptor, FcεRI- or FcεRII (CD23)-bound IgE, or FcγR -bound IgG. Examination of these possibilities is highly warranted.
HRF AS A SUPERANTIGEN OR AUTOANTIGEN
The broad reactivity of HRF with the Fab portion of Igs (~30% of Igs) and its ability to activate mast cells indicate that HRF can be considered a 'mast cell superantigen'. In addition to typical superantigens (such as staphylococcal enterotoxin B), which can bind to MHC II and a substantial subset of T cell receptors, there are B cell superantigens (such as Staphylococcal protein A, Peptostreptococcus magnus protein L, and HIV gp120), which target large clonal sets of B cells. B cell superantigens interact with the conserved antigen receptor variable (V)-region sites, which are different from antigen-binding sites.42 Some B cell superantigens can also activate mast cells and/or basophils by interacting with FcεRI-bound IgE.43-45 However, unlike the B cell superantigens, the HRF-binding sites at least partially overlap with antigen-binding sites. Our sequence analysis of a limited number of IgE and IgG molecules indicated that HRF-reactive Igs use specific V gene families of κ (kappa) light chain.46 The exact binding sites must be analyzed through structural studies.
More recently, we found that IgEs' binding of autoantigens correlates with differences in HC versus PC properties.47 Most mouse monoclonal HC IgEs exhibited polyreactivity to double-stranded (ds) DNA, single-stranded (ss) DNA, β-galactosidase, thyroglobulin, and/or HRF. In contrast, mouse PC IgEs failed to react with these antigens. A human monoclonal HC IgE also displayed polyreactivity to HRF, dsDNA, and ssDNA. Interestingly, sera from AD patients showed increased reactivity to ssDNA and β-galactosidase and increased levels of HRF. Some AD patients, but not healthy individuals, had substantial serum levels of HRF-reactive IgE. Sera from AD patients with high titers of DNA-reactive IgE could induce several fold more IL-8 secretion in human mast cells than could sera from healthy individuals. These results show that most HC, but not PC, IgEs exhibit polyreactivity to autoantigens, supporting the autoimmune mechanism in the pathogenesis of AD. It has been thought that autoimmunity underlies allergic diseases.48 Consistent with this notion, the prevalence of allergies and autoimmune diseases has increased in parallel.49,50 Recent studies support the concept that IgE autoreactivity may play a pathogenic role in severe and chronic forms of atopy.48,51 Acute allergic reactions following exposure to exogenous allergens can be understood as immediate-type inflammation triggered by degranulation of mast cells via allergen-IgE-FcεRI interactions, whereas chronic allergic inflammation with Th1 characteristics can occur and persist in the absence of exogenous allergens. IgE-mediated presentation of autoallergens may activate autoreactive Th1 cells to release proinflammatory cytokines.52,53 Several environmental allergens share striking structural and immunologic similarities with human proteins.54-58 A large number (≥140 proteins) of IgE-reactive autoantigens were found in AD patients, and some of them were shown to have the abili-ty to activate basophils.59 Therefore, the autoimmune mechanism could be an important part of the disease pathogenesis in a subset of AD patients.
FUTURE PERSPECTIVE
Our studies imply that elevated levels of HRF and/or HRF-reactive Igs activate mast cells and basophils and amplify inflammation.46,47 Although it is unknown whether HRF can activate mast cells and basophils under homeostatic conditions (or physiological levels and the repertoire of IgE and IgG), HRF-mediated mast cell activation should contribute to the amplification of allergic reactions when IgE, IgG, and HRF levels are high enough at the allergic inflammatory site. It is noteworthy that there is a level of HRF in normal serum that does not induce noticeable inflammation. Thus, there should be a mechanism(s) by which normal levels of HRF do not harm the animal. As a relatively large fraction (~30%) of Igs can bind HRF, changes in the antigen repertoire of Igs may cause a change in the levels of HRF-reactive Igs. Thus, it will be interesting to investigate the potential involvement of HRF in other pathological conditions such as autoimmune or infectious diseases. Overall, recent studies encourage us to further understand the mechanism by which HRF plays a proinflammatory role in allergy and other diseases. As human IgE also can interact with human HRF, attempts to translate the most recent observations to human pathology will be a major focus in the future, raising hope for improving the arsenal with which we combat allergic diseases.

 XML Download
XML Download