

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Recently, there has been a dramatic increase in the prevalence of noncommunicable diseases (NCDs), such as cardiovascular disease, metabolic disease, cancer, chronic obstructive pulmonary disease, inflammatory bowel diseases, and allergic diseases.1 A key driver of the development of NCDs, including allergic diseases is chronic low-grade inflammation, which participates in the pathogenesis of both metabolic and immune-mediated diseases.2 The metabolic and immune systems may also regulate each other through sharing of cellular machinery: such inter-relationships may contribute to the development of NCDs.3
Environmental factors may play a key role in the pathogenesis of NCD as they can promote the development of chronic low-grade inflammation by altering immune responses. Repeated exposure to such environmental stimuli over the course of a life-time may strongly promote the development of NCDs, especially if there is an imbalance between the inflammatory responses and the host response against environmental toxins.4 Indeed, there is considerable evidence that indicates that effective immune responses have been disturbed by recent changes in environmental factors such as diet, microbial colonization patterns, air pollution, and climate, as a result, the prevalence of NCDs has increased.5,6
According to the developmental origins of health and disease hypothesis, exposure to environmental factors such as nutritional state and tobacco smoke in utero shapes the body structures, function, and metabolism of the offspring and thereby affects their development of NCDs later in life.7 Several studies have shown that exposure to environmental factors during pregnancy also alters the immune system in offspring, partially through epigenetic dysregulation and gene-environment interactions.7 However, further studies are needed to evaluate the effect of environmental factors during fetal periods on the health of the offspring in later life.
In this review, we will elucidate the effect of environmental changes and gene-environment interactions on the development of allergic diseases. We also hope that by demonstrating the effect of environmental factors on the development of allergic disease, this review will spur research into the protective strategies for allergic diseases.
Environmental risk factors of allergic diseases
The rapid increase in the prevalence of allergic diseases and other NCDs within a relatively short period can be attributed to environmental changes over the last few decades, as particular changes in dietary patterns, climate change, environmental pollution, and factors such as mode of delivery that shape microbial colonization patterns.6,8,9 These factors have been shown to promote the initiation and persistence of allergic and other inflammatory diseases through chronic low-grade inflammatory responses.10,11
With regard to diet, several studies showed that Westernized diets, are partially responsible for the increased prevalence of allergic disease. They are characterized by increased consumption of ω-6 fatty acids found in margarine and vegetable oils and decreased consumption of ω-3 fatty acids found in fish oil and vegetables.11,12 The effect of dietary fatty acids on the development of allergic diseases is due to anti-inflammatory effects of ω-3 fatty acid and pro-inflammatory effect of ω-6 fatty acid.12 Decreased intake of antioxidants such as vitamins C and E has also been associated with a higher risk of wheeze and reduced pulmonary function.13 The effect of nutrition on allergic diseases may be mediated by immunomodulation,14 oxidative stress,15 and epigenetic mechanisms.5
Many observational studies have shown that climate changes are associated with the increased prevalence and severity of allergic diseases.9 Furthermore, environmental pollutants such as ozone, particulate matter (PM), and nitrogen dioxide (NO2) have been implicated in the recent rise in the incidence of allergic diseases.16 Although the mechanisms underlying these associations have not been fully elucidated, some of the possible theories are discussed below.
Several recent studies showed that mode of delivery, antibiotic use, or diet changes affect the pattern of microbial colonization.17,18 The changes in the intestinal microbiota in particular may shape the mucosal immune system in the gut and lungs and thereby promote allergenic mucosal immune responses.17,19 Our previous study supported this by showing that oral administration of Lactobacillus rhamnosus ameliorates the asthmatic response in mice.20
The more pro-inflammatory Westernized diets and environmental pollutants, along with clean environments have also been shown to influence the composition and functions of the gut and airway microbiota.21,22 The developing immune system is shaped by the different responses of the host microbiota to environmental changes; it is also influenced by changes in the composition of microbiota. These interactions among the microbiota, host, and environment can induce the low-grade inflammation that promotes the development of allergic diseases.22
Environmental factor: Climate
Climate change is defined by the Intergovernmental Panel on Climate Change as a "change in the state of the climate that can be identified by changes in the mean and/or the variability of its properties and that persists for an extended period, typically decades or longer. It refers to any change in climate over time, whether due to natural variability or as a result of human activity". Although climate change is an ongoing process, it has received particular attention recently, in part because there is evidence that global warming has affected human health adversely. The average temperature of the Earth's near-surface air and oceans has increased by 0.74±0.18℃ over the past century.23 This has caused the world to experience not only more hot days and rainy days with consequent flooding but simultaneously also more frost days and drought periods. Thus, climate change imposes adverse effects directly by increasing the intensity of extreme weather events such as floods and droughts.
Pollen and mold are major inhalant allergens that can promote allergic diseases. Climate change affects the levels and distribution of these allergens. For example, raised temperature increases pollen production, extends the pollination period, and allows the spread of particular plants to larger areas, thereby causing more individuals to be more exposed to them.24 In addition, the high carbon dioxide (CO2) levels produced in urban areas can augment the allergenicity of pollen by elevating its allergenic compound levels.25 The increased frequency of thunderstorms caused by climate change is also associated with outbreaks of asthma that is mediated by bursts of pollens or fungal spora.26 Moreover, the rise in sea level and the changes in rainfall patterns increase the frequency of mold allergies.27 In addition, modern buildings, which are designed to be air-tight to save energy, will probably increase the indoor humidity, thereby worsening mold allergies.
Climate change can also augment the adverse effects of air pollution by shaping its distribution and preventing its dispersal. Moreover, raised temperature can elevate the levels of secondary air pollutants such as ozone at ground level. Furthermore, even stable levels of ozone or PM can synergize with high temperatures in causing adverse health effects.28 Thus, the greenhouse effect may potentiate the multiple interactions between various air pollutants.
Environmental factor: Microbiota
Microflora hypothesis
The hygiene hypothesis was proposed more than 20 years ago by Strachan to explain the dramatic increase in the prevalence of allergic diseases over the last few decades. In its original form, this hypothesis claimed that limiting early-life infections impedes the development of the natural immune system and thereby predisposes individuals to allergic disease.29 The modified "microflora hypothesis" proposes that the overlying hygienic Western lifestyle limits not only infections, but also general microbial exposure and it alters the colonization of the human gut, which in turn disrupts the development of immune systems and ultimately leads to allergic diseases.18,30,31 Infancy and early childhood have been identified as important and vulnerable periods in the development of the gut microbiota, which shapes the disposition to allergic diseases.
The importance of microbial exposure during the perinatal period has been studied in animal models. It is difficult to achieve oral tolerance in germ-free mice because they lack Th1 responses and have stronger Th2 responses, as demonstrated by the increased production of IL-4.32 Administration of lipopolysaccharides together with food antigens in mice increased the tolerance of foods.33 In addition, the proper development of oral tolerance seems to require a complex intestinal microbiota rather than colonization with a single microorganism.34 These results suggested that the gut microbiota is a key factor that shapes the development and regulation of the immune system early in life.
After birth, the diversity of the gut microbiota increases with age.35 Since healthy human fetuses are thought to develop within a bacteria-free environment, the nature of the first contact of the neonate with the germ-containing environment may markedly shape the early microbiota composition. Indeed, the mode of delivery has been shown to influence the early colonization pattern in neonates: vaginally delivered infants acquire bacterial communities that resemble the microbiota of their mother's birth canal, which are dominated by Lactobacillus, Prevotella, or Sneathia spp., whereas, infants born by Cesarean section harbor bacterial communities that are similar to those found on the skin surface and are dominated by Staphylococcus, Corynebacterium, and Propionibacterium spp.18,36 A meta-analysis also showed that Cesarean delivery can be a risk factor for allergic rhinitis, asthma, hospitalization for asthma, and perhaps food allergy/food sensitization, but not for inhalant sensitization or atopic dermatitis.37 Moreover, the gut flora composition of atopic infants is similar to that of infants born by Cesarean section.38 Other factors have also been associated with allergy, such as probiotics use and antibiotic use in neonates and infants that alter the gut micriobiota. A Korean birth cohort study (The COhort for Childhood Origin of Asthma and allergic disease, COCOA) showed that Cesarean delivery and prenatal antibiotic exposure may affect the gut microbiota, which may in turn influence the risk of atopic dermatitis in infants.18
To confirm the pathogenesis of allergic diseases shaped by microbiota composition, it is necessary to perform prospective studies which show the different population of microbiota, and function of immune system in allergic and non-allergic infants. Moreover, only a few prospective studies have used metagenomic analysis to examine the association between gut microbiome and allergic diseases. Two of these showed that low microbial diversity early in life is associated with an increased risk of allergic diseases.39,40 One study showed that reduced gut bacterial diversity in infants associates with an increased risk of allergic sensitization, allergic rhinitis, and peripheral blood eosinophilia in the first 6 years of life.39 These results support the general hypothesis that imbalances of gut microbiota influence the development of allergic diseases.
Biodiversity in the environment
The hygiene hypothesis also suggests that microbial biodiversity in the environment may influence our immune system and health later in life. Supporting this is the fact that microbial-rich environments confer protection against allergic diseases41 and poor biodiversity is associated with human immune dysfunction. For example, the homes of atopic individuals have significantly lower environmental biodiversity than those of healthy adolescents.42 The atopic adolescents also have significantly less diverse gram-negative gammaproteobacteria on their skin. Furthermore, the abundance of the genus Acinetobacteria on the skin correlates positively with the expression of IL-10 in peripheral mononuclear cell. These observations suggest that the Western lifestyle and living in urban areas hampers the exposure to microbes that is needed to develop a healthy microbiota, which in turn is essential for the proper development of immune tolerance and homeostasis.
However, the relationship between environmental and human microbiota remains unclear. Repeated skin and fecal measurements in 2 subjects showed that only a small fraction of all taxa found within a single body site are present across all time points.43 This suggests that the human core microbiota is small and that many taxa are present on some but not all occasions. While it seems plausible that the composition of the human microbiota can be temporarily changed by external influences, such as inhalation or ingestion of microbiota and that farm- or animal-associated microbiota may compete with the microbiota that promote asthma, it remains unclear whether such exposure can permanently change the colonization patterns.
Connections between human microbiota and systemic immunity
The above studies do not exclude the possibility that the microbiota composition is influenced by the immune system rather than the other way around. However, there are several lines of evidence that changes in the human microbiota can lead to different stable immune states.44,45 Commensal microbiota functions in the immunoregulatory network and the Toll-like receptor (TLR) system. The development and maintenance of epithelial-cell integrity, tolerance, and tissue repair requires the interaction with microbes and immune cells via their specific receptors. When these microbial stimuli are absent, it cannot adequately induce the immunoregulatory circuits including regulatory dendritic cells, regulatory T (Treg) cells and regulatory cytokines such as IL-10, transforming growth factor-β (TGF-β).46 Interestingly, it has also been shown that an inflammatory milieu enhances the conversion of Treg cells to inflammatory Th17 cells and enriches the bacteria that tolerate to the inflammatory mediators in microbiota, thereby creating a self-perpetuating system. These molecular findings further support the hygiene hypothesis, which states that a sedentary lifestyle in affluent urban environments does not provide adequate microbial exposure for the development of a healthy microbiota.
Epigenetic mechanisms have also received considerable attention as a possible explanation as to how environmental exposure modulates the immune system. In general, microbe-rich environments induce both pro-inflammatory and immunoregulatory circuits early in life, which indicates an early activation of the relevant genes.47
The importance of microbial contact during the crucial periods of fetal life, delivery, and infancy for the healthy immune system and metabolic programming is only now being understood. This understanding creates new opportunities for improving infant's health and reducing the risk of disease in later life (Fig. 1). However, the underlying mechanisms, the cause-effect relationship, and particularly the crucial individual microbes and microbial compounds still have to be elucidated.
Environmental factor: Air pollution
Outdoor air pollution
Air pollution has been markedly increased by the rapid growth of the world's motor vehicle fleet along with recent urbanization. Motor vehicles emit large quantities of noxious pollutant including CO2, carbon monoxide (CO), hydrocarbons (HC), nitrogen oxides (NOx), and PM. Traffic-related air pollution (TRAP), including these primary air pollutants and secondary by-product such O3, can have adverse effects on health and the environment. Among these pollutants, particle pollution and ground-level ozone are the most widespread threats to human health.
PMs are major pollutants along with gaseous pollutants such as NOx and O3. They consist of variously sized particles but the smaller particles have greater adverse health effects because they can penetrate the lung as well as the blood stream more easily. Diesel exhaust particles (DEPs) emitted from motor vehicles are major sources of airborne PM and account for up to 90% of the airborne PM in urban areas. DEPs are known to generating reactive oxidative stress (ROS).
Ozone is a secondary air pollutant that is produced by photochemical reaction of primary pollutants such as NOx and volatile organic compounds (VOCs) under ultraviolet light. NO2 is generated by most combustion processes that use air as the oxidant. Since the most prominent sources of NO2 are the internal combustion engines of automobiles, NO2 levels may be an indicator of TRAP. In addition, NO2 and other nitrogen oxides also contribute to the generation of ozone and other oxidant pollutants. It is difficult to determine the independent effect of NO2 on human health in epidemiological studies because NO2 is often seen together with other pollutants derived from combustion exhaust and it acts as a precursor for secondary pollutants. Nevertheless, several studies have shown that long-term exposure to NO2 can decrease lung function and increase respiratory symptoms and it is also associated with immunological impairments.
Air pollution combined with exercise exacerbates allergic asthma and decreases the lung function in asthmatics.48 Air pollution also causes asthma and increases airway hyperresponsiveness.49 Interestingly, it may act synergistically with host susceptibility (as indicated by past episodes of bronchiolitis) to promote the development of asthma.50 Allergic rhinitis is also closely associated with air pollution. The prevalence of allergic rhinitis is higher in populations that reside in more polluted areas or near roads with heavier traffic than in populations with less pollutant exposure.16,51 While air pollution is considered to be a respiratory environmental factor because it is primarily inhaled into respiratory systems, it is also associated with eczema and skin disease.52 Indeed, a recent study has shown that the amount of air pollutants such as NO, toluene, and VOCs correlate with eczema symptoms.53 Although there is a great deal of consistent evidence that shows positive associations between air pollution and the development of allergic symptoms, it is still controversial whether air pollution can cause allergic sensitization.54 Recent human and animal studies showed that air pollutants such as ozone and DEPs increase the prevalence of allergic sensitization and stimulate the production of allergen-specific IgE antibodies, respectively.16,55
Lastly, in East Asia, Asian sand dust that typically occur in early spring have been receiving increased attention due to their significant ecological and health effects, contribution to the aggravation of pre-existing allergic diseases, and augmentation of harmful effects by air pollution.
Future studies using relevant biomarkers reflecting exposure to air pollutants and the consequent effects on target organs will give more information about how to cope with air pollution. Moreover, while the independent effect of each air pollutant should be determined, it is also important to assess their effects when they are combined with other environmental or host factors.
Indoor air pollution
The effect of indoor air pollution should also be assessed closely. Indoor air pollutant concentrations can be 2- to 5-fold higher than outdoor levels and most people spend as much as 90% of their time indoors.56 Such chronic exposure to indoor pollutants at home or in the work-place warrants careful assessment of their health impacts. The sources of indoor air pollution include the combustion of fuel for heating the home and cooking, tobacco smoke, chemical products used for cleaning, building and decorating, and dust from outside.
The indoor air pollution in residences, day-care centers, retirement homes and other special environments affects specific population groups that are particularly vulnerable due to their health status or age. Moreover, dampness is more likely to occur in modern buildings, which are crowded and lack appropriate ventilation, may have higher levels of microbial pollution that involve hundreds of species of bacteria and fungi that grow indoors easily. Exposure to such microbial contaminants is associated clinically with respiratory symptoms, allergies, asthma and immunological reactions.57
Environmental tobacco smoke (ETS) has been identified as a major source of indoor air pollution.58 Several studies showed that especially fetal and infantile exposure to tobacco smoke increased the risk of asthma symptoms and lower respiratory tract infection in early childhood.59,60 ETS exposure also increases the vulnerability of the lungs to other air pollutants such as PM.61 Additionally, a recent study showed that prenatal exposure to cockroach allergen increased the risk of allergic sensitization in children at the age of 5-7 years and that exposure to nonvolatile polyaromatic hydrocarbons (PAHs) augmented the risk.62 Moreover, VOCs are emitted from interior products such as synthetically coated furniture, carpets, and polyvinylchloride flooring, which means that newly built or renovated houses have higher levels of these compounds. Children have been shown to have increased levels of pro-inflammatory cytokines such as TNF-α and IL-6 after indoor activities63 and exposure to VOCs is associated with respiratory symptoms and allergic manifestations through increased inflammation as a result of oxidative stress.64
Other environmental chemical pollutants
Modern people are often exposed to a variety of chemical pollutants as well as increased levels of air pollution at home or in the workplace during their daily life. Heavy metals such as arsenic and nickel are widespread environmental chemicals that are associated with various adverse health effects, including cancer, cardiovascular diseases, neurological disease, and autoimmune disease. They exert their deleterious effects by inducing ROS, which in turn cause the oxidative deterioration of biomolecules or epigenetic changes.65 Pesticides and various chlorination byproducts in drinking water have also been shown to induce epigenetic alterations that are associated with an increased risk of cancer.66,67 However, the effect of these environmental chemicals on allergic disease has been poorly studied to date.
Possible links between environmental change and allergic disease
Oxidative stress
Oxidative stress plays a central role in the mechanisms by which air pollutants damage human health. Many air pollutants exert their deleterious effects by inducing oxidative stress in the cells and tissues with which they have contact. In particular, TRAPs are known to form ROS such as superoxide anions, hydrogen peroxide, and hydroxyl radicals. Oxidative stress arises when ROS overwhelms antioxidant defenses. After this imbalance is reached, ROS react readily with proteins, lipids, and DNA, resulting in a number of pathological consequences (Fig. 2). A primary consequence of oxidative stress is the lipid peroxidation or the oxidative degeneration of lipids. If this process is not stopped, this reaction can lead to cell death through damaging cell membranes. Activation of signaling pathways is another way by which oxidant pollutants may induce pathological responses in the lung. Given these observations, it is likely that individual susceptibility to air pollution may depend on genetic variation in the antioxidant system. Indeed, common genetic variants of the glutathione-S-transferases, which play a role in cellular protection against oxidative stress,68 are associated with the allergic diseases in a general population with exposure to air pollution. As for air pollution, NO2, PM2.5, PM10, and ozone were often estimated.56,58 Genetic polymorphisms of the antioxidant system and their interactions with environmental factors will be discussed later.
Epigenetics
It has been the subject of considerable research that environmental factors, especially air pollution, are able to induce epigenetic changes that promote allergic diseases. These studies are summarized in Table 1. The studies in humans showed that PM decreases the degree of methylation of the iNOS promoter, which is involved in defense to oxidative stress.69,70 An animal study also showed that DEP challenge induces the hypermethylation of IFN-γ and the hypomethylation of IL-4, thereby skewing the immune system towards Th2 responses.71
The epigenetic effect of smoking also has been investigated in children. Cigarette smoking can affect the development of asthma by inducing global and gene-specific methylation or by modifying histone protein.72,73,74 Interestingly, regardless of maternal smoking, the smoking of grandmothers associates significantly with asthma in their grandchildren at the age of 5.75 This suggests that there is transgenerational inheritance of epigenetic changes. In addition, in utero exposure to traffic-related PAH induces a specific methylation mark that serves as a potential surrogate marker for the subsequent development of asthma in offspring.76 Besides the epigenetic effects by air pollution, an animal study that assessed the protective effect of farm exposure on asthma revealed that maternal exposure to farm-derived gram-negative bacteria protected the offspring from asthma by preventing the reduction of histone 4 acetylation within the IFN-γ promoter.77 In general, low early-life microbial exposure is associated with hypermethylation of the IFN-γ gene of naïve T cells, which is in turn associated with an increased risk of allergy (Fig. 3).78 These observations call for more research on the role of microbial stimuli in the epigenetic modulation of T cells, particularly Treg cell function.
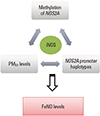
Not only environmental factor but also genetic variation can affect or interact with the epigenetic pattern. DNA methylation of the filaggrin gene worsens the effect of heterozygosity of loss-of-function variants on eczema.79 A recent remarkable study also demonstrated three-way interactions among DNA genetic variations, epigenetic variations, and air pollution in terms of phenotype expression (Fig. 4).69 These findings suggest that the modification of epigenetic pattern by genetic variation may further interact with environmental factors, thus resulting in phenotypic alterations.
In terms of methodology, epigenetic changes are dependent on specific tissues or cell types and they may be generated in a time-dependent manner. For these reasons, it is recommended that studies on asthma-related epigenetics involve several measurements over time and that the most appropriate target cells are selected. Of particular use in such research will be birth cohorts, where pregnant women are enrolled and followed up. This will allow the epigenetic modifications that occur during the critical period of life to be identified as well as the life-long effects of these modifications. Since epigenetic changes can pass into the next generation, such epigenetics research may shed more light on the most effective methods of preventing allergic disease.
Gene-environmental interaction for allergic disease
The concept of gene-environmental (GxE) interactions has evolved in the last century and has now become a central theme in studies to assess the causes of allergic disease.80 Although many studies have been performed to search for genes associated with allergic disease, genes alone cannot explain the recent increase of allergic disease. Another explanation is that genes act in association with environmental change to develop the allergic diseases. Technological advances have greatly influenced the recent burst of research into GxE interactions in allergic diseases.81 These studies have led to the identification of novel associations of genetic variants with disease, the improved measurement of environmental factors by developing exposure biology tools, and the development of new methods for identifying GxE interactions (Table 2).82,83,84,85,86,87,88,89,90,91,92,93
With regard to asthma, the most well-known and extensively studied GxE interactions are those between (1) the CD14 gene and endotoxin exposure,94 (2) HLA genes and allergens,95 and (3) TLR genes and infectious agents.96 Recent genome-wide environment interaction studies (GEWIS) on asthma also discovered interactions between genome-wide single nucleotide polymorphisms and farm-related exposure in Central Europe82 and smoking exposure.97
Given the increasing evidence showing that air pollution promotes allergic diseases, many studies have investigated the interplay between genetic susceptibility and air pollution on allergic diseases. Since air pollution induces oxidative stress and thereby promotes inflammation, most of these studies focused on genes in the oxidative stress and inflammatory pathways.
The antioxidant system is a complex network that is composed of many enzymes, of which glutathione S-transferase (GST) enzymes play a particularly important role. They are essential for glutathione homeostasis, which is a determinant of the cellular response to oxidative stress and cytoprotection from the byproducts of oxidative stress. The GSTM1 and GSTP1, sub-classes of GST, have been studied most frequently. These studies assessed how the polymorphisms in these enzymes modify individual susceptibility to air pollution and promote its deleterious effects. However, there is some debate about whether children who inherit a Val (105) variant allele in GSTM1 are protected from a higher risk of asthma with exercise, especially in high ozone levels.98 The inconsistency of the results suggests that gene to gene interactions (i.e., interactions between GSTP1 and other antioxidant genes) and epigenetic mechanisms should be considered along with GxE interactions.99
A previously described study showed that PAH augmented allergic sensitization in offspring with prenatal exposure to cockroach allergen, especially in children with GSTM1 null mutations.62 These findings suggest that in utero exposure may predetermine the physiological and metabolic adaptations that are made in later life, especially in genetically susceptible individual.
Several recent studies that detected GxE interactions examined diverse environmental determinants, particularly smoking, farming, indoor and occupational environments, air pollution, and diet or nutrition.82,83,84,85,86,87,88,89 To improve our understanding of the GxE influences on allergic diseases, newly identified genetic and environmental factors should be studied. The latter factors include microbiota and stress, especially during the prenatal or early postnatal period. Future studies should also clearly define the outcomes (phenotypes) of allergic diseases. In addition, such studies should consider various times of the GxE interaction because so many studies show that timing of exposure has crucial effects on susceptibility to disease due to lung growth, immunological maturation, and epigenetic alterations.80 Longitudinal studies with multidisplinary research teams are also needed to assess the effect of the environment on allergic diseases properly.
CONCLUSIONS
The increasing prevalence of allergic diseases is attributable to environmental changes such as the Westernized diet, air pollution, climate changes, and other factors that influence the environmental and host microbiota. These environmental changes are associated with the loss of microbial biodiversity, a reduced human microbiota, immune dysregulation, and subsequent low-grade chronic inflammation, which is a common pathogenic mechanism of NCDs, including allergic diseases.
Environmental micro-organisms, previously unique and abundantly present, are key players in the induction and maintenance of immunoregulatory circuits and tolerance. The composition and dynamics of our microbial communities and the innate and adaptive immunity are affected by our lifestyle and environment, constantly. Changes in climate and lifestyle as well as air pollution have the potential to influence changes in the microbiome. More prospective and experimental studies are required in regard to the understanding of human and environmental microbiome for the prevention and reduction of allergic diseases.
In addition, early life period including in utero is a critical period to shape the later life phenotype, especially through the epigenetic mechanism. Therefore, for establishing the strategies to prevent allergic disease, there is a need to elucidate early environmental effects on the epigenetic changes related with allergic diseases in later life and to identify genetically susceptible individuals. In this regard, large-scale birth cohort studies including diverse environmental changes that assess the effects of genetic and epigenetic modifications during the critical periods of life and their life-long effects of these modifications are warranted.





 XML Download
XML Download