

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Atopic dermatitis (AD) is one of the most common chronic inflammatory skin diseases, characterized by xerosis, eczematous skin lesions, pruritus, immunodysregulation, epidermal barrier dysfunction and IgE-mediated sensitization to food and environmental allergens.1 Our understanding of the disease pathogenesis of AD has increased, and it is clear that no single defect accounts for the array of clinical features associated with AD. Lifetime prevalence of AD varies from approximately 8% to 18% worldwide.2 AD is on the rise in many developing countries, affecting over 10% of the population, and has reached a plateau of ~20% in Western countries.3,4 The striking increase in AD incidence observed in recent decades has been attributed to the resettlement of populations from rural to urban areas. According to the hygiene hypothesis,5 a lack of early childhood exposure to a variety of microbes results in reduced immune tolerance.
Of the various pathogenic factors suggested for AD, impaired epidermal permeability barrier function is considered to be important. Many studies have suggested that a defective epidermal barrier combined with an abnormal immune response might contribute to the pathophysiology of AD, supporting the outside-inside hypothesis.6,7 In addition, the clinical manifestations of AD predate the development of asthma and allergic rhinitis, suggesting that AD is an entry point for subsequent allergic disease in a process called the atopic march. The concept of atopic march was hypothesized to describe the progression of atopic disorders from AD in infants to asthma and allergic rhinitis in children. Kubo et al.1 suggested a theoretical model of barrier disruption followed by percutaneous sensitization, which may apply to the pathogenesis of the atopic march. Therefore, the importance of barrier disruption in AD has gained increasing attention. In this review, we discuss the recent progress in understanding the functions of the epidermal permeability barrier, its immunologic role in human and animal subjects, and barrier repair therapies in AD.
Epidermal permeability barrier dysfunctions in AD
The skin, as an interface between the organism and the external environment, plays a major role in protecting and supporting the life it encloses. The outermost layer of the skin, the stratum corneum (SC), is the primary mediator of this epidermal permeability barrier function.8 Atopic dry skin displays impaired barrier function, indicated by an increased transepidermal water loss (TEWL) and decreased water-binding capacity due mainly to altered levels of inter/intracellular components in the SC.
Filaggrin and its gene
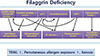
Filaggrin (FLG) is named after the filament aggregating protein, and the FLG gene is located on chromosome 1q21 within the epidermal differentiation complex.9 FLG protein is localized in the stratum granulosum (SG). Profilaggrin, a 400-kDa polyprotein, is the main component of keratohyaline granules. In the process of keratinocyte differentiation, profilaggrin is dephosphorylated and cleaved into 10-12 FLG molecules. In vitro, FLG monomers aggregate and align keratin bundles, which contribute to the mechanical strength and integrity of the SC.6 To promote corneocyte compaction, FLG monomers are degraded into their constituent amino acids, including glutamine, arginine and histidine. These amino acids are then hydrolyzed further into acidic polycarboxylic acid osmolytes, which maintain SC hydration (the so-called natural moisturizing factors, NMFs), by caspase 14, peptidylarginine deiminases or bleomycin hydrolase to maintain hydration of the upper SC and to reduce skin surface pH.5,10 Maintaining acidic pH is key for many protective functions, including permeability barrier homeostasis, SC integrity and cohesion, antimicrobial defense, and it is important for the activation of enzymes involved in ceramide metabolism and modulation of the serine protease cascade required for coordinated epidermal differentiation and cornified cell envelope formation.6,11 Moreover, FLGs are crucial proteins for terminal differentiation of epidermal keratinocytes, which form a skin barrier with the SC intercellular lipids. Pyrrolidone carboxylic acid, a major component of NMFs in skin, is primarily produced from FLG proteins in the SC and plays a vital role in maintaining hydration of the SC.8 If these proteins are decreased or absent due to FLG mutations, the FLG-associated SC barrier is disrupted. Such SC barrier disruption results in decreased formation and secretion of the lamellar body (LB), cornified envelope, and corneodesmosome, elevated pH and decreased tight junction (TJ) proteins, leading to increased episodes of percutaneous allergen exposure (Fig. 1).1 Kezic et al.12 confirmed that individuals with FLG-null mutations have significantly reduced levels of NMFs in the SC of their forearms and palms. Moreover, significantly lower NMF levels were observed in individuals with a history of AD who were carriers of FLG mutations compared with those who were non-carriers. The authors demonstrated higher TEWL in the carriers of FLG mutations compared with non-carriers. FLG has been suggested to contribute to the formation of acid mantle (described below) within the SC through the production of urocanic acid (UCA) via the filaggrin-histidine-UCA cascade.13 Consequently, FLG-deficiency in AD lesions leads to defects in the formation of the cornified envelope, a decreased ability to maintain SC hydration, and a parallel elevation in pH. The increase in pH enhances the KLK5 and KLK7 activities, which are optimal at neutral pH, resulting in over-degradation of corneodesmosomes and decreased SC integrity and cohesion. Patients with FLG mutations might have a higher risk of allergic sensitization compared with those with wild-type FLG.14 FLG mutations are significantly correlated with increased risk of developing atopic diseases, including AD, atopic asthma, allergic rhinitis and nickel and food allergies, even though FLG is not found in the bronchial epithelium.15
Nevertheless, FLG null mutations may not be sufficient to induce the findings typical of AD. The median prevalence of FLG mutations among Europeans and Asians is 7.7% and 3.0%, respectively.16 In European studies, the prevalence of FLG mutations in AD subjects is 3% in Italy, 15.2%-22.9% in Germany, 40.2%-42% in the UK and 45.2%-55.8% in Ireland, and this pattern suggests a higher tendency for FLG mutation prevalence in AD patients who reside in countries of higher latitudes than in those of lower latitudes.16 Patients with FLG mutations can develop ichthyosis vulgaris without manifestations of AD, and approximately 50%-90% of AD patients have no FLG defects.16,17 In fact, mice with the FLG null mutation exhibited no spontaneous AD-like skin lesions.18 However, flaky tail mice homozygous for the 5303delA mutation in the FLG and Tmem79/Matt genes showed FLG and NMF deficiencies, presumably representing the characteristics of AD after exposure to allergens.19 A recent study revealed that the matted gene Matt is a predisposing gene for AD in mice, and a common Matt (Pl use consistent designations when describing flaky tail mice in this section and later in the manuscript) single nucleotide polymorphism is associated with AD in human subjects.19 In addition, Sasaki et al.20 showed that the Tmem79 (ma/ma) mutation is responsible for the spontaneous dermatitis phenotype in matted mice, probably due to the impairment of the lamellar granule secretory system and altered SC barrier function, which has also been identified in Caucasians with FLG mutations in a dose-dependent manner.21 A recent study by Kim et al. showed that IL-25 inhibits the expression of FLG and acts synergistically with Th2 cytokines to inhibit FLG expression.22 We assume that the barrier breakage in AD is due not only to the FLG gene alone, but that FLG combined with other genes might cause or exacerbate AD skin lesions.
TJ barrier
TJs are involved in the control of paracellular migration of inflammatory cells. Increasing evidence shows that skin TJs, which localize in the SG, contribute to epidermal barrier formation,23 however, the role of TJs in AD is still unknown. Among TJ proteins, claudin (Cldn)-1, a TJ-specific integral membrane protein, and Cldn-4 play important roles in the barrier function of the skin. In a recent study, Cldn-1 knockout mice demonstrated normal SC structure but abnormal SC formation and SC barrier defects. These Cldn-1 knockout mice died shortly after birth and had increased TEWL.24 In humans, a lack of Cldn-1 causes NISCH syndrome, which is characterized by ichthyosis, scalp hypotrichosis, scarring alopecia, neonatal sclerosing cholangitis and leukocyte vacuolization. Kubo et al.25 reported that dendrites of activated Langerhans cells express Cldn-1 and ZO-1. Therefore, functional bicellular and tricellular TJs, which can take up external allergens easily when the SC barrier is disrupted, are present between Langerhans cells and keratinocytes.1 In a recent study, patients with AD demonstrated a defect in TJ barrier function and composition.26 Furthermore, Yuki et al.27 reported that impaired TJ barriers affect polar lipids and profilaggrin processing by disturbing the pH of the SC. Based on these reports, we assume that decreased TJ components in lesional AD promote epicutaneous penetration of allergens, including the house dust mite antigen, which leads to the increased possibility of developing systemic allergy or atopic march. Recently, Kuo et al.28 showed that the activation of toll-like receptor (TLR)-2, which shows reduced expressed in AD, enhances TJ function in mice and human keratinocytes, suggesting the use of TLR-2 enhancers as a potential therapeutic strategy in patients with AD. Further investigation of the crosstalk between TJs and the SC barrier is needed to clarify the role of TJs in the pathogenesis of AD.
PH, proteases and protease activated receptor (PAR)2
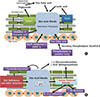
The skin surface pH, which ranges from 4.5 to 5.5 in humans, is slightly acidic compared with the normal physiologic pH.8 The importance of pH in vivo was first reported in experiments in which the permeability barrier function was disrupted acutely, producing a parallel increase in pH.29 The pH of the SC influences key epidermal functions, including permeability barrier homeostasis, desquamation of corneocytes, initiation of inflammation, processing of secreted LB polar lipids and antimicrobial defense (Fig. 2A). The UCA breakdown products, free fatty acids and sodium hydrogen antiporter (NHE1) are the three main factors responsible for maintaining acidic pH in the skin.9 In AD, the baseline skin pH is more alkaline than the average baseline pH of healthy skin.30 The perturbation of lipid metabolism and its molecular organization as well as elevated skin pH induce the growth of bacteria such as Staphylococcus aureus (S. aureus).
Trans-UCA (t-UCA) is an endogenous acidifier of the SC. Hence, decreased production of FLG products could result in an initial increase in skin surface pH that is sufficient to activate multiple serine proteases in SC, which exhibit neutral-to-alkaline pH optima.31 If prolonged, the pH-induced increase in serine protease activity could precipitate downstream structural and functional alterations, as mentioned below (Fig. 2B). Moreover, increased pH leads to higher activities of Kallikrein (KLK) 5 and 7 (SC chymotryptic enzyme), which have optimal activity at neutral pH, resulting in the over-degradation of corneodesmosomes and a decrease in SC integrity and cohesion.13 Moreover, elevated skin pH inhibits the activity of lipid-processing enzymes, including β-glucocerebrosidase (β-GlcCer'ase), acid sphingomyelinase (aSMase) and secretory phospholipase A2 (sPLA2), which are essential in the production of ceramide and free fatty acids, thereby causing impaired lipid processing and defects in the epidermal permeability barrier.
SC acidity is important in antimicrobial barrier function in that it inhibits the growth of pathogens. The growth of S. aureus, which readily colonizes in the lesional skin of AD, is normally inhibited at low skin pH and grows best at pH 7.5. In AD, the increased pH in lesional skin leads to bacterial growth, resulting in allergic inflammation and aggravation of AD. Hatano et al.11 showed that the acidification of SC alone substantially prevents the development of barrier abnormalities and downstream immune abnormalities during the elicitation phase in the murine oxazolone-induced AD model. This study suggests that the maintenance of a normal or hyperacidic pH could be a possible novel treatment for reversing or preventing AD in humans.
PAR is a G protein-coupled receptor, characterized by a unique mechanism of self-activation following specific proteolytic cleavage of its extracellular domain.13 Four PAR members have been identified: PAR-1, -3, and -4 are activated by thrombin and are involved in homeostasis and thrombosis, whereas PAR-2 is activated by trypsin-like serine proteases, but not by thrombin. PAR-2 is widely distributed throughout the mammalian body13 and is expressed by almost all skin cell types, especially keratinocytes. Previous studies demonstrated that PAR-2 is expressed in the suprabasal epidermal layers of both humans and mice, and that this expression is most prominent in the granular layer, implying that PAR-2 expression might depend on the state of epidermal differentiation.32 PAR-2 localizes in the suprabasal layers of the mouse and human epidermis with high levels in the SG, supporting PAR-2 as the primary sensor of barrier-initiated serine protease activity.33 The lesional skin of AD expresses high levels of PAR-2. Furthermore, PAR-2 activation is likely to be involved in pruritus of AD.13 Indeed, high expression rates of PAR-2 and trypsin have been observed in the lesional skin of patients with AD, and PAR-2 agonist peptides induce pruritus in patients with AD.34 PAR-2 in the skin is involved in pruritus via the transient receptor potential vanilloid receptor (TRPV1) inflammation, and thymic stromal lymphopoietin (TSLP) activation, implying that PAR-2 antagonists represent potentially useful therapeutics for treating AD (Fig. 3).35 The association between PAR-2 and TSLP is discussed below.
Many studies have provided evidence that genes associated with proteases/protease inhibitors become deregulated in patients with AD, shifting the balance between proteases and protease inhibitors toward increased protease activity. A link has been demonstrated between AD and a 4-base pair (AACC) insertion into the 3'-untranslated region of the KLK7 gene, which increases the half-life of KLK7 mRNA and the enzymatic activity of KLK7.13 Lymphoepithelial Kazal-type-related inhibitor (LEKTI) is an inhibitor of multiple serine proteases in skin, and it regulates the enzymatic activities of serine proteases, including KLK5, -6, -7, -13, and -14, thereby controlling epidermal barrier function.36 A premature stop codon mutation in the SPINK5 gene, which encodes LEKTI, is associated with Netherton syndrome (NS), a rare ichthyosiform dermatosis characterized by congenital ichthyosiform erythroderma, severe atopic manifestations and hair shaft abnormalities. SPINK5-deficient mice demonstrate increased proteolytic activities of KLK5 and KLK7 in the epidermis and abnormal degradation of desmoglein 1, resulting in abnormal corneodesmosome cleavage and premature desquamation. These data suggest that LEKTI is a key regulator of KLK5 and KLK7 activity, and defective SC adhesion caused by epidermal protease hyperactivity is the primary pathogenic event in NS.13 In addition, several studies reported that the polymorphisms in the SPINK5 gene are associated with AD.37,38 Moreover, cystatin A, a cysteine protease inhibitor, inhibits the endogenous cathepsins B, -H, and -L and the exogenous proteases from house dust mites of dermatophagoides pteronyssinus (Der p) 1 and dermatophagoides farinae (Der f) 1.13,39 The cystatin A-encoding gene (CSTA) polymorphism (+344c variant) is associated with AD.39 This CSTA variant causes reduced levels of cystatin A in the skin surface and in sweat, allowing exogenous proteases to break down the integrity of the SC, which enhances the penetration of allergens and aggravates AD. These genetic variants in protease and protease inhibitor genes coexist in some patients with AD, resulting in a more severe defect in the skin barrier.13
The proteolytic activity from various contact allergens and aeroallergens are considered important factors in the initiation and aggravation of AD. Proteolytic activity plays a role in the pathogenesis of allergic diseases, including allergic rhinitis, asthma and AD, by inducing Th2 allergic inflammation and directly affecting the structure and function of the epidermal barrier, which facilitates further penetration of allergens through the defective skin barrier.40 House dust mite and cockroach allergens, important environmental factors in the pathogenesis of AD, have been shown to have proteolytic activity.41 House dust mite allergens contain several cysteine and serine proteases. The serine proteases from the mite allergens, Der p 3 and Der p 9, activate PAR-2 on keratinocytes to produce cytokines and contribute to the pathogenesis of AD, whereas the cysteine protease activity in Der p 1 stimulates inflammation via a PAR-2-independent mechanism. S. aureus produce extracellular V8 protease, which causes defective epidermal permeability barrier function in the skin of hairless mice by directly degrading DSG1.42 These reports imply that proteolytically active allergens could break down the skin barrier via PAR-2-mediated inhibition of LB secretion or PAR-2-non-mediated mechanisms, including the degradation of corneodesmosomal proteins and lipid processing enzymes. This degradation further triggers allergen penetration through the disrupted epithelial barrier to aggravate Th2-mediated inflammation and possibly causes the non-IgE-associated form of AD (atopiform AD) to switch to the typical IgE-associated form of AD.13 Various roles of serine proteases and PAR-2 are described in Fig. 3.
Peroxisome proliferator-activated receptors (PPARs)
PPARs are ligand-activated transcription factors involved in the genetic regulation of lipid metabolism and energy homeostasis. PPARs belong to a subfamily of nuclear hormone receptors comprising three different isoforms of PPARs: PPARα, PPARβ/δ and PPARγ.43 After ligand binding, PPARs can regulate gene expression by binding to peroxisome proliferator response elements in target genes, after heterodimerizing with the retinoid X receptors. Activators of PPARs and liver X receptors display potent but largely positive effects on the epidermal structure and function in normal and diseased skin, including the upregulation of FLG, anti-inflammatory activity, and reverse epidermal hyperplasia and differentiation.44 In AD, PPARs reduce several inflammatory mediators in the skin and regulate epidermal barrier homeostasis by stimulating epidermal differentiation and lipid production. In fact, PPAR ligands inhibit T helper cell responses by inhibiting IL-2 production in T cell clones.45 Since patients with AD exhibit primary abnormalities in the epidermal barrier function, the PPAR/LXR activators might possess potential utility in AD by stimulating epidermal differentiation and lipid production.44 Lower expression of PPARα is observed in AD lesional skin, suggesting that decreased PPARα signaling might be involved in the relationship between permeability barrier abrogation and allergic inflammation in AD.46 Experiments using PPARα knockout mice revealed a modest decrease in the epidermal expression of differentiation markers (profilaggrin and loricrin), a thinner SG than that in wild type mice, decreased keratohyaline granules, normal permeability barrier function and decreased β-GlcCer'ase activity.47 Indeed, the combined therapy of topical PPARα activators/ligands and steroids showed not only potent anti-inflammatory benefits in the murine AD model, but also almost no rebound flares, which is one of the considerable side effects of steroid therapy.48 Furthermore, PPARα prevents the side effects of steroid-induced structural and functional abnormalities and improves paracellular permeability.48 Recent analysis of AD skin lesions showed increased PPARγ expression in keratinocytes and in infiltrating T cells and monocytes.49 PPARγ plays a critical role in the regulation of genes involved in cellular proliferation, in specific components of the Th2 inflammatory pathway and in maintenance of the skin barrier.45 Systemic treatment with a PPARγ agonist, rather than with topical treatment, decreased the amount of surface area affected, the severity of lesions and the number of flares in patients with severe AD.50 Therefore, PPARs and their corresponding ligands are potential therapeutic targets for treating AD. Further studies are needed to verify the underlying mechanism of PPARs in AD pathogenesis.
Ceramides and lipid processing
The major lipid species of the SC are ceramides, fatty acids and cholesterol. The majority of SC lipids are derived from the LB and synthesized in the keratinocytes of the upper stratum spinosum and SG.8 In human AD, a selective reduction in ceramide content, especially ceramide 1, is observed. Indeed, Nakajima et al.51 reported that a ceramide deficiency in the epidermis leads to psoriasis-like lesions in the serine palmitoyltransferase knockout mouse model. The abnormal neutral skin pH in patients with AD inhibits the activity of lipid-processing enzymes, including β-GlcCer'ase, aSMase and sPLA2, thereby causing impaired lipid processing and a defective lipid barrier, as mentioned above. Other minor mechanisms that might account for the frequently colonized microbial pathogens in AD involve acidic ceramidase activity, which could decrease ceramide content.52 Moreover, altered ceramide metabolism in AD results in decreased levels of sphingosine, which is produced from ceramide by ceramidase in the SC. Since sphingosine is known to have a powerful antimicrobial activity on S. aureus at physiologic levels, downregulation of sphingosine is one explanation for the susceptibility of patients with AD to S. aureus colonization.37,38
Antimicrobial peptides
Antimicrobial peptides (AMPs) are important molecules involved in the evolutionarily conserved innate immune defense system of the skin against bacteria, viruses, fungi, parasites and tumor cells.53 More than 1,700 AMPs have been identified.54 In normal human skin, keratinocytes are the main source of AMPs, and AMP synthesis occurs primarily in the SG.53 In addition to their antimicrobial activity, AMPs in the skin have variable functions, including modulation of host inflammatory responses and promotion of wound healing.55 Moreover, AMPs have been shown to be associated with permeability barrier function in a knockout mouse model for cathelin-related antimicrobial peptide (CRAMP), a murine homologue of LL-37. These knockout mice showed a significant delay in permeability barrier recovery after tape stripping as well as structural abnormalities in LB content.56 In addition, a recent study suggested that human β-defensin-2 (HBD2) can be used as a marker for disease severity and barrier properties in AD.53
AD skin has significantly lower AMP production than does psoriatic skin, while both disorders are associated with inflammation and defective barrier function.57 Nomura et al.58 proposed that this defect in AD is caused by AMP suppression via elevated levels of T-helper 2 cytokines, IL-4 and IL-13. Lower AMP levels diminished the antimicrobial barrier function in AD subjects, resulting in increased susceptibility to S. aureus and other microbial superinfections, such as eczema herpeticum.53,59 In fact, LL-37 exhibited antiviral activity against herpes simplex virus (HSV), and mice deficient in the LL-37 homologue showed higher levels of HSV-2 replication in their skin. Moreover, the skin of AD subjects with eczema herpeticum exhibited significantly lower levels of LL-37 than that of AD patients. Therefore, reduced levels of LL-37 may render AD subjects susceptible to eczema herpeticum.59,60
Many reports have demonstrated that AMPs are induced in the skin in AD. Reduced gene expression of hBD-3 was not observed in the epidermis of patients with AD compared with psoriasis,60 and increased expression levels of hBD-3 and other AMPs, such as hBD-2, psoriasin, elafin and calprotectin, were found in AD skin compared with healthy skin.61 However, another study did not detect differences in hBD-2 and LL-37 expression levels between the nonlesional skin of AD patients and non-AD patients, indicating that expression of hBD-2 and LL-37 is not decreased at baseline in the epidermis of patients with AD.62 Ballardini et al.63 and Gambichler et al.64 detected higher levels of AMPs in lesional skin than in nonlesional skin in AD. Based on these contradictory reports, we assume that the increased AMP amounts are lower at the onset of inflammation in the lesional skin in AD patients compared with that in non-AD patients. Further studies are needed to clarify the causality of AMP expression on lesional and non-lesional epidermis in AD.
Ultraviolet irradiation and AD
Ultraviolet (UV) C radiation is absorbed by the ozone layer; however, the skin is constantly exposed to UVA and UVB irradiation, and this UV radiation affects FLG degradation. Using histidine as a substrate, histidase produces t-UCA, a major UVB-absorbing epidermal chromophore. t-UCA is photoisomerized to cis-UCA with UVB exposure, and cis-UCA may produce oxidative DNA damage and initiate the translation of genes associated with apoptosis and immunosuppression.16,65 Although UV irradiation of the skin induces disruption of the epidermal barrier, phototherapy is generally used in patients with AD to relieve acute flares (UVA1) and chronic lesions (narrow band UVB).66 The mechanism of action in AD has not been elucidated; however, the proper UV dose has positive effects on the epidermal permeability barrier function by immunomodulating apoptosis of inflammatory cells, reducing the number of cutaneous nerve fibers, inhibiting the Langerhans cells and altering cytokine production.67 In addition, a suberythemal dose of UVB on the skin exerts beneficial effects on the SC barrier function by activating the cutaneous vitamin D system.68,69 In addition, UV initiates endoplasmic reticulum (ER) stress responses including downstream activation of the transcription factor, NF-κB.70 In the epidermis, ER stress increases human cathelicidin antimicrobial peptide (CAMP) expression, which stimulates innate immunity.71 A recent study suggested that UV radiation enhances cortisol activity in a waveband-dependent manner, and cortisol production protects and/or restores epidermal barrier homeostasis.72 In fact, most patients affected by AD improve during the sunny summer season, and artificial UV radiation, including psoralen-UVA and narrow-band UVB, is frequently utilized in the treatment of AD. Further comparative clinical studies are needed to clarify the proper dose and wavelength of UV therapy in AD.
Link between the epidermal permeability barrier function and the systemic immune reaction in ad: thymic stromal
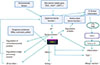
TSLP is an interleukin-7-like cytokine produced by epithelial cells that triggers dendritic cells to induce Th2 inflammatory responses.73 TSLP enhances the effector stage of the allergic response and amplifies secretion of Th2 cytokines in synergy with IL-1 and TNF-α.74 Furthermore, TSLP is induced by exogenous stimuli, such as defects in barrier differentiation, infections, TLR ligation and trauma, as well as by topical application of active vitamin D3 and low-calcemic analogue treatment.75 TSLP is overexpressed in keratinocytes in AD skin lesions and triggers a pathway underlying AD progression and atopic march. In addition to early protection from skin barrier defects in AD, the selective inhibition of TSLP might be a key intervention strategy to block the progression of atopic march. Previous studies have reported increased levels of TSLP in tissues biopsied from acute and chronic AD skin lesions and from the serum of patients with AD.76 Overexpression of TSLP in epidermal keratinocytes is a systemic driver of bronchial hyperresponsiveness, and TSLP in asthmatic airway epithelial cells is correlated with reduced lung function.77 The PAR-2-TSLP relationship, in particular, has been emphasized in many studies.35 Indeed, PAR-2 might be a parent signal that induces the overexpression of TSLP in epithelial cells.78 A recent study revealed high TSLP expression when keratinocytes of flaky tail mice were exposed to a PAR-2 agonist, but low TSLP expression in the presence of a PAR-2 antagonist, suggesting that PAR-2 is at least partly responsible for the induction of TSLP production.79 These results suggest that TSLP could be a link between skin barrier function and the systemic immune reaction in AD.
Animal models in AD
Many research groups have established animal models of AD. In this section, we discuss several AD animal models that have been used most frequently and which seem to correlate with the outside-inside hypothesis in AD. Flaky tail mice have been used to investigate the role of FLG in AD. The spontaneous flaky tail (ft) mouse arose on the background of an existing recessive hair phenotype, matted (ma). Flaky tail mice have a 1-bp frameshift mutation in the FLG gene. Flgft mice have spontaneous dermatitis with increased IgE levels, endogenous protease (KLK5, 7, and 14) activity, TSLP expression, and basophil accumulation in the skin, showing many characteristics of AD.79 They appear normal at birth, and the flaky phenotype becomes visible 3 days after birth with the presence of dry scaly skin and tail constrictions. Under specific pathogen-free conditions, the majority of Flgft mice developed clinical and histological eczematous skin lesions similar to human AD with outside-inside skin barrier dysfunction, as evaluated by newly devised methods.80 Moreover, the topical application of dermatophagoides pteronyssinus in Flgft mice induces skin lesions that are clinically and histologically similar to those seen in AD.79 The relative contribution of Flg and ma to the compound phenotype has yet to be defined fully. As mentioned above, the matted mouse gene is a predisposing gene for AD, and the Tmem79 (ma/ma) mutation is responsible for the spontaneous dermatitis phenotype in matted mice, which can also be found in Caucasians with FLG mutations in a dose-dependent manner.21 Based on these reports, the flaky tail mouse is perhaps the most represented animal model of AD corresponding with FLG mutation in human AD.
Oxazolone (Ox) is commonly used to provoke allergic contact dermatitis, and it evokes a Th1-dominated response to the hapten.81 However, multiple challenges of Ox to the skin of hairless mice over an extended period cause the skin inflammation to shift from a Th1-dominated delayed type hypersensitivity response to a chronic Th2-dominated inflammatory response similar to human AD.11,44,48,81,82 Nine to 10 Ox challenges to hairless mice produced a chronic Th2-like skin inflammation, which was characterized by dermal infiltration of Th2 lymphocytes (expressing CRTH2), mast cells and eosinophils, increased expression of IL-4 in the dermis and highly elevated IgE levels. Repeated Ox challenges led to increased epidermal hyperplasia and decreased expression of keratinocyte differentiation markers. Skin barrier abnormalities were associated with increased TEWL. Additionally, impaired LB secretion along with decreased SC ceramide content and hydration resulted in reduced lamellar membranes, similar to those observed in patients with AD. Furthermore, as in human AD, increased epidermal serine protease activity in the SC and decreased expression of 2 LB-derived antimicrobial peptides (CRAMP and mBD3) paralleled the decrease of their human homologues in AD skin lesions. Although the repeated hapten sensitization model is not genetically driven, this model may be applicable to the acquired form of AD with respect to extrinsic allergens. Because of its reproducibility, predictability, low cost, and relative rapidity, the repeated Ox sensitization model could be useful for evaluating pathomechanisms and potential treatments for AD.
Nc/Nga mice, an inbred mouse strain reported by Matsuda et al., provided the first model of murine AD.83 Nc/Nga mice have mutations on chromosome 9 linked to increased IgE production and Th2 responses. Skin lesions develop spontaneously in Nc/Nga mice after exposure to various exogenous aeroallergens, closely mimicking human AD. In addition, Nc/Nga mice show skin barrier abnormalities with increased TEWL and abnormal skin conductivity under conventional conditions and impaired ceramide metabolism.84 However, Yagi et al.85 suggested that AD-like skin changes in Nc/Nga mice are IgE/Th2-independent because STAT6-deficient Nc/Nga mice exhibit skin changes, comparable to those of STAT6-positive Nc/Nga littermates, and undetectable IgE serum levels. Therefore, the Th2-mediated immune response is not necessary for the development of AD-like skin disease in Nc/Nga mice.86
Several studies have employed the occlusive patch of ovalbumin (OVA) to generate AD and allergic march models. Although asthma-like lung lesions developed in these models, AD-like skin lesions, including eczematous lesions, and disrupted SC barrier function did not fully develop. Zhu et al.87 used IL-13 transgenic mice treated with OVA. Demehri et al.88 used RBP-jCKO mice treated with OVA, and Hershko et al.89 used Ox challenge with OVA treatment. Although all of these models expressed AD-like skin lesions combined with asthma-like lung lesions, the methods relied on intraperitoneal injections of OVA for sensitization, not epicutaneous sensitization via damaged skin barriers. We suggest that the allergic march model correlating with the outside-inside hypothesis might be generated using topically applied allergens (such as house dust mites) onto AD-like skin lesions of Flgft mice, Ox-induced AD model mice and Nc/Nga mice with disrupted barriers. In addition, inhalation of the allergens might reveal physiologic mechanisms of allergen penetration, systemic circulation, and allergen challenges.
Barrier repair therapy in AD
Although anti-inflammatory treatments might reduce the disease severity by suppressing the immune reaction, they do not fully correct the primary underlying barrier abnormalities that drive disease pathogenesis in AD. The ideal emollients for barrier repair therapy should normalize the epidermal barrier function by reducing TEWL, improving SC hydration and protecting the skin from allergen penetration. AD treatment must aim to restore the barrier and its function, as well as suppress inflammation and control the pruritus. In addition, it is important to educate patients by highlighting the chronic nature of AD, the importance of maintenance therapy and the need for prompt suppression of flare-ups.90 General considerations and approaches to treating AD by restoring the barrier system include education (about soaps, hydration and lowering stress levels), hydration (using emollients to decrease the use of steroids), decreasing S. aureus carriage, breaking the itch-scratch cycle use of antihistamines, which are also helpful for barrier function,91 reduction of pH in the SC (hyperacidification),11 and use of epidermal barrier-enhancing agents, including serine protease inhibitors, topical PAR2 antagonists, PPAR and LXR activators, AMP enhancing agents and TJ component replacement therapies, some of which need further clinical research to clarify the treatment effect on AD.
Moisturizers have a pivotal role in improving and maintaining the skin-barrier function and in reducing skin susceptibility to irritants.92 Properties of physiologic lipid-based products differ from nonphysiologic agents. Compared with other emollients that form a more superficial occlusive barrier (e.g., petrolatum), ceramide-dominant physiologic lipid-based products are thought to participate in physiologic lipid replacement therapy by permeating the SC, incorporating into keratinocyte synthesis, becoming processed in the LBs and being secreted back into the SC.93 The ceramide-dominant, triple-physiologic lipid barrier repair therapy for AD (Cer:cholesterol:free fatty acids at a 3:1:1 molar ratio) addresses the problems of both a global reduction in lipids and the further decline in Cer in AD.52,94 Studies on AD and barrier repair treatment showed that adequate lipid replacement therapy reduces inflammation and restores epidermal barrier function comparable to that by topical fluticasone cream.95,96 Due to the decreased amount of ceramide in AD, the approach of ceramide supplementation was introduced using pseudoceramide, which showed a close similarity to that of SC intercellular lipids. The physiologic lipid vehicle mixture containing pseudoceramide showed positive results in an AD model.97,98 Hatano et al.48 reported that steroid therapy combined with PPARα ligand therapy is not only effective, but it also prevents the development of steroid-induced side effects, thereby reducing the amount of steroid required in a murine AD model. These studies imply that barrier repair therapy reduces the usage and side effects of steroid, which enhances the efficacy of AD treatment. These studies suggest the theoretical possibility that the combined treatment of calcineurin inhibitors and barrier repair therapy might be a useful modality for AD treatment (Table 1). Further clinical studies involving the combined treatment of steroid and/or other proactive therapies and barrier repair therapies are needed. Urea, used in various topical emollients, has been shown to restore barrier function (transglutaminase-1, involucrin, loricrin, FLG and epidermal lipid synthetic enzymes) and AMP expression (LL-37 and HBD2) by regulating epidermal gene expression in a murine AD model.99 This study suggests the potential use of urea in therapeutic applications for AD treatment. Histamine could aggravate AD via its effects on epidermal keratinocyte differentiation. In a recent study using an animal model of AD, Gschwandtner et al.100 reported that histamine inhibits epidermal keratinocyte differentiation and skin barrier formation in organotypic skin models. The ability of topical histamine type 1 and 2 receptor (H1/2r) antagonists to target epidermal H1/2r could translate into increased efficacy in the treatment of inflammatory dermatoses, likely due to decreased inflammation and enhanced barrier function by stimulating the synthesis and secretion of epidermal lipids.91 Therefore, H1r and H2r antagonists could be used as a topical barrier repair therapy to treat AD.
The anti-inflammatory features of barrier repair therapy could be explained by several mechanisms. Normalizing the barrier function induces decreased cytokine cascade, prevention of allergen and/or hapten ingress and increased antimicrobial defense. Furthermore, certain free fatty acids have anti-inflammatory properties. In addition, normalizing the pH using barrier repair therapy leads to downregulation of serine protease activity, which causes reduced Th2 inflammation, IL-1 activation and PAR2-mediated pruritus. Regarding the pathomechanisms described above, barrier repair therapy for AD should include a prolonged pH reduction in the SC (hyperacidification) and the application of serine protease inhibitors, topical PAR2 antagonists, PPAR and LXR activators, AMP-enhancing agents or replacement of TJ components. Further studies are needed to develop novel and effective barrier repair therapies for AD on the basis of AD pathogenesis.
CONCLUSIONS
The epidermal permeability barrier plays a crucial role in maintaining skin homeostasis. Perturbations in barrier function have significant effects on overall skin quality. The barrier anomalies facilitate sustained antigen ingress through the defective barrier, which can bring about a Th2-dominant response. It enhances the TEWL, resulting in dry skin and leading to the release of preformed proinflammatory cytokines and to a cascade of inflammatory events. These events negatively affect the epidermal permeability barrier function, which causes a vicious cycle in the skin, perhaps leading to systemic inflammation. The converging pathogenic features described above provide a basis for the development of specific strategies to restore the barrier function in AD.
The defective epidermal barrier in AD could allow the so-called atopic march, characterized by the epicutaneous delivery of antigens that induce asthma and allergic rhinitis. Although FLG is not expressed in either bronchial or other non-keratinizing mucosal epithelia, FLG deficiency is associated with mucosal atopy independent of AD. Based on this observation, barrier repair therapy might block the development of atopic march.

 XML Download
XML Download