

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Exclusion of smokers from asthma efficacy trials
In the pursuit of minimizing potential bias, randomized controlled trials (RCTs) typically exclude patients with characteristics that may obscure or diminish the measureable efficacy signal. The highly-selective nature of respiratory RCTs, and more specifically of asthma RCTs, can result in the exclusion of more than 95% of the general patient population.1,2 Thus, although registration RCTs are rigorous and provide the data necessary to establish the efficacy of new interventions (in idealized patients and practice conditions), they are limited in the extent to which their results can be extrapolated to reflect potential treatment effects at the population level.
As there are many factors that can interfere with efficacy, RCTs are designed with necessarily strict inclusion/exclusion criteria and require highly-interventional (and standardized) clinical management regimens.3,4 RCT patients are intensively monitored throughout clinical trials to ensure complete compliance with the prescribed regimen, to allow measurement of different markers and parameters, and to ensure early detection of possible treatment-related adverse events. In contrast, routine care asthma patients tend to be prescribed therapy then sent home in the belief (or hope) that they will follow their prescribing instructions and will remember how to perform the appropriate inhalation maneuvers. Patient - clinician interactions in routine care are typically driven by necessity - a need for additional therapy or when there is a problem to report.
While intensive RCT clinical monitoring aims to minimize areas of potential bias, tight patient selection is also necessary. RCT recruitment often includes only patients with no (or negligible) comorbid illnesses or concurrent medications, those with good inhaler technique and high adherence to study therapies; a clear-cut asthma diagnosis and some degree of lung function impairment; substantial reversibility to short-acting beta-agonists and frequent rescue medication usage. Lifestyle characteristics such, as cigarette smoking, typically result in patient exclusion. Indeed RCT inclusion criteria around smoking are growing increasingly tight, often resulting in exclusion of not only current smokers, but also any patients with a history of 10 pack years or more.
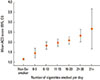
The rationale for excluding smokers from asthma trials is understandable in light of the knowledge that active smoking in asthma is associated with worsening symptoms, accelerated decline in lung function, and impaired response to corticosteroids.3,5,6 The association between steroid resistance and cigarette exposure was nicely demonstrated in a recent UK study designed to evaluate the association between self-report smoking status (as well as rhinitis and medication adherence) and asthma control. A total of 3,916 patients from 85 different UK primary care practices completed anonymised questionnaires capturing validated asthma control data (the Asthma Control Questionnaire [ACQ]). Compared to never smokers, current smokers had a greater than 4-fold increase (odds ratio [OR]=4.33; 95% Confidence interval [CI]: 3.58-5.23; P<0.001) in risk of poorly-controlled asthma (ACQ >1.5). Although not as marked as for current smokers, ex-smokers also had significantly increased odds of having poorly-controlled asthma (OR=1.59; 95% CI: 1.36-1.87; P<0.001) compared with never smokers. Moreover, there was a clear relationship between the number of cigarettes smoked each day and asthma control (F[5,655]=6.08, P<0.001; Fig. 1).3
These findings were echoed in a randomized, placebo-controlled study in mild asthma that compared the effect of inhaled fluticasone propionate (1,000 µg daily) or placebo over 3 weeks in 38 steroid-naive adult asthma patients (17 smokers; 21 non-smokers). Efficacy was assessed using morning and evening peak expiratory flow (PEF) readings, spirometric parameters, bronchial hyper-reactivity, and sputum eosinophil counts. After 3 weeks of fluticasone therapy, mean morning PEF reduced for current smokers while there was a significantly increase for non-smokers (-5 L/min vs 27 L/min, respectively). While non-smokers receiving fluticasone experienced improvements across all endpoints compared with placebo, no differences in outcomes were observed for current smokers receiving fluticasone therapy compared with those on placebo. These data suggest that in mild asthma, active cigarette smoking impairs the efficacy of short-term ICS treatment - an important consideration when managing mild asthma patients who smoke.6
Mechanisms of steroid resistance in smokers
There are a number of mechanisms and processes thought to contribute to the diminished response to ICS treatment seen in asthma patients who smoke.
Histone deacetylase 2 impairment
The molecular mechanism by which corticosteroids switch off the expression of inflammatory genes in diseases such as asthma is relatively well understood.7,8 In chronic inflammation pro-inflammatory transcription factors activate coordinated expression of multiple inflammatory genes (including cytokines, chemokines, adhesion molecules, and inflammatory enzymes). The increase in gene expression is brought about by acetylation of the core histones around which DNA is wound in the chromosome, as a result of the intrinsic histone acetyltransferase activity of co-activator molecules to which these transcription factors bind.9 This unwinding of DNA allows RNA polymerase II to bind and initiate transcription that results in increased synthesis of inflammatory proteins. Corticosteroids reverse the process by deacetylating the hyperacetylated histones through the recruitment of histone deacetylase 2 (HDAC2) to the activated coactivator complex.9 The process results in rewinding and compaction of DNA, exclusion of RNA polymerase, and suppression of inflammatory gene transcription. This mechanism is thought to account for the anti-inflammatory effect of corticosteroids in asthma. Impairment of HDAC2 activity can, therefore, lead to a pronounced reduction in responsiveness to corticosteroids.
Oxidative stress
Although corticosteroid therapy has become the backbone of asthma management approaches, its utility in chronic obstructive pulmonary disease (COPD) is less clear. Although asthma and COPD both involve inflammation of the respiratory tract, the nature of the inflammation typically differs as COPD is normally characterized by a predominance of neutrophilic inflammation and high levels of oxidative stress that result in impaired HDAC2 activity, while asthma is usually characterized by eosinophilic inflammation and lower levels of oxidative stress.10,11 However severe8,10 and refractory asthma12 are also known to be characterized by increased neutrophils, and neutrophil levels in the sputum of asthma smokers have also been shown to be elevated compared to those of asthma patients with no smoking history.13 An increase in neutrophils and macrophages is the innate immune responses to cigarette smoke-induced tissue damage and the resultant increase in oxidative stress could account for the reduced corticosteroid responsiveness seen in smokers.8,14,15
Increased cysteinyl leukotrienes production
Cigarette smoking also stimulates the production of cysteinyl leukotrienes (CysLTs) possibly through COX-1 induction - a group of inflammatory mediators (leukotrienes C4, D4, and E4) central to asthma pathophysiology. They are associated with microvascular permeability and mucus hypersecretion and are potent smooth muscle constriction agents.16,17 Leukotriene E4 (LTE4) is a stable end product of CysLTs metabolism in the human lung. Urine LTE4 is a reliable marker of endogenous CysLTs formation and can, thus, be used to monitor changes in the rate of CysLTs production.18 Gaki et al. conducted a study of 40 asthma patients (20 smokers) to investigate whether smoking significantly affects urine LTE4 levels. The authors found significantly higher LTE4 levels in asthmatic smokers (164[48] pg/mg) compared with non-smokers (87[26.3] pg/mg; P<0.0001), which may suggest potential differences underlying CysLTs inflammatory processes in asthmatic smokers.13
Small airway remodeling
Another feature of cigarette smoking that has been implicated in poor response to traditional inhaled corticosteroid therapy is the greater presence of small airway disease in smokers. For patients with obstructive lung disease, exposure to cigarette smoking is associated with infiltration of inflammatory cells into the small airways of the lung (typically peripheral or distal airways of ≤2 mm in diameter). Smoking also induces growth factor production in the airway walls and results greater small airway remodelling. The consequence of this process is a marked increase in airflow resistance in the small airways and functional evidence of chronic airflow obstruction.15-22 If the small airways do play an important role in the pathogenesis of asthma in smokers, the limited effect of traditional ICS therapies may be associated with their failure to penetrate the small airways and address a key site of asthmatic inflammation.15
Management options
Although it is generally accepted that cigarette smoking is associated with diminished steroid response, asthma guidelines provide no specific management advice for smokers.23 When faced with a smoking asthma patient in the clinic, physicians must piece together the available evidence to make prudent and judicious prescribing decisions drawing on the limited evidence available. A short review of the available data for different asthma management approaches in smokers follows.
Smoking cessation
In terms of management, the first treatment recommendation for smoking asthma patients should be smoking cessation. A 6-week asthma study showed cessation resulted in a significant improvement in forced expiratory volume in 1 second (FEV1) (P=0.040), a significant reduction in sputum neutrophils (P=0.039) and improvement in total cutaneous vasoconstrictor response score to topical beclomethasone compared with continued smoking over the same period.5,24 Furthermore, in a study on 147 patients with asthma (66 never-smokers, 46 exsmokers, and 35 current smokers) replicating these clinical results, the epithelial characteristics in ex-smokers turned out to be similar to those in never-smokers, suggesting that the smoke-induced tissue changes can be reversed by smoking cessation.25
However, while cessation can be advocated by the clinician, it is not always welcomed by the patient and there will continue to be substantial numbers of smoking asthma patients whose therapeutic management requires additional consideration due to the attenuating effects of smoking on corticosteroid efficacy.
High-dose prescribing or dose escalation
Although several studies have demonstrated that cigarette smokers with asthma are insensitive to short-term ICS, data are limited on the possibilities of different ICS dosing strategies. To explore these issues, patients with mild asthma (n=95) were recruited to a multi-centre, randomized, double-blind, parallel group study comparing inhaled beclomethasone dipropionate (BDP) at doses of 400 µg or 2,000 µg daily in smokers and non-smokers over a longer, 12-week, period. The primary endpoint was the change in morning PEF. Secondary endpoints included evening PEF, use of reliever inhaler, number of asthma exacerbations, spirometric parameters, and asthma control score. After 12 weeks of inhaled BDP therapy, there was a considerable difference in morning PEF of smokers compared with non-smokers with asthma (P=0.035). Interestingly, the difference between smokers and non-smokers was less pronounced for patients in the 2,000 µg BDP arm (-15 [-50 to 21], P=0.40) compared with that seen in the 400 µg BDP arm (-25 [95% CI -45 to 24]; P=0.019) (Fig. 2). Similarly, while there was a significantly higher exacerbation rate among smokers compared to non-smokers in the 400 µg BDP treatment arm (6 vs 1, respectively, P=0.007), no significant difference was seen in exacerbation rates in patients receiving the high 2,000 µg BDP daily dose (1 vs 2, smokers vs non-smokers, respectively, P=0.661).26 These data build on the results of shorter-term studies showing diminished ICS efficacy in mild smoking asthmatics treated on low-dose ICS, by suggesting not only that diminished ICS efficacy holds true over a longer treatment course, but also that the disparity between treatment response in smokers and non-smokers appears to be reduced at higher ICS doses. Thus use of ICS therapy at higher dose may be a valid treatment consideration in patients with a current smoking status.5,26
Leukotriene receptor antagonist therapy
Cysteinyl leukotrienes play an important role in the pathogenesis of asthma, rhinitis (allergic and non-allergic), exercise-induced asthma, aspirin-intolerant asthma, and virus-triggered asthma exacerbations. Combining this knowledge with Gaki et al's reported elevation of urine LTE4 concentration in smoking asthmatics compared with non-smoking asthma patients, it could be hypothesized that 'targeting leukotriene' could be a solution in smoking asthmatics.13,24
A large, randomized, double-blind, parallel-group study was recently conducted across 31 countries to evaluate the efficacy and safety of montelukast (10 mg once daily) and high-dose inhaled fluticasone (250 µg twice daily) compared with placebo in smoking asthmatics (self-reported smoking of 0.5-2 packs a day).27 It was the largest asthma trial involving smokers conducted to date and randomized 347, 336, and 336 patients to montelukast, fluticasone, and placebo, respectively. Over the 6-month outcome period, patients in both the fluticasone and montelukast treatment arms demonstrated significantly more days of asthma control (44.97% vs 39.05%, P=0.04). Significant improvements on the secondary endpoint of change from baseline in mean daytime symptom score were observed or both fluticasone and montelukast compared with placebo (P=0.004, P=0.001, respectively); there were no significant differences between montelukast and fluticasone (P=0.321) (Fig. 3). However, fluticasone did significantly improve the change from baseline in mean morning PEF compared with placebo (P≤0.001) while montelukast had no significant effect (P=0.117). Smoking history and exposure appeared to play a role in response to therapy - patients who had a smoking history of less than 11 pack years tended to show more benefit from fluticasone while those with a smoking history of more than 11 pack years tended to show greater numerical benefit with montelukast. Overall, these data suggest that montelukast could be new treatment option to high-dose inhaled corticosteroid for asthma patients who choose to continue smoking.
Extra-fine particle corticosteroid therapy
Based on the rationale that small airway abnormalities appear to contribute to the pathogenesis of asthma in patients who smoke, new ICS therapies that generate particles small enough to penetrate the small airways may be more effective than traditional ICS options.15
While traditional dry powder inhalers or chlorofluorocarbon-metered dose inhalers generate particles with a median mass aerodynamic diameter of 2-4 µm,28 newer MDIs using hydrofluoroalkane (HFA) as the propellant can generate an aerosol of smaller particles with a median mass aerodynamic diameter of approximately 1 µm.29 Studies have shown that these extra-fine particle ICS formulations have a lung deposition rate of 50%-60% and penetrate more deeply into the peripheral airways than drugs delivered via traditional inhalers.30,31 Thus, it has been hypothesized that targeting inflammation in both the central and peripheral airways may be beneficial, particularly for asthma phenotypes where small airways involvement is particularly implicated.15
This hypothesis was tested in an observational asthma study using clinical records pooled form the United Kingdom's Clinical Practice Research Database32 and Optimum Patient Care Research Database.33 The study was designed to explored the real-life effectiveness of extra-fine HFA BDP and standard particle size fluticasone delivered via MDI devices across patients of different smoking subgroups: non-smokers and in current and ex-smokers (combined). Using database measures of asthma control and exacerbations, current/ex smokers prescribed extra-fine HFA-BDP had significantly lower exacerbation rates and significantly higher odds of achieving asthma control than patients prescribed fluticasone. Although exacerbation rates were also lower and odds of control higher among non-smokers prescribed extrafine HFA-BDP compared to those receiving fluticasone, the difference was less marked than in the current/ex smokers cohort.34 Although further data from studies of complementary study are required, these observational study data suggest targeting the small airways with an extra-fine formulation ICS may offer benefits over traditional ICS formulations in asthmatic smokers.
Combination therapy
Data are limited on the potential utility of combination therapy in smoking asthma patients. The long acting beta2-agonists formoterol and salmeterol have been shown to restore the glucocorticoid-dependent transactivation suppresed by smoke in vitro35 and, in a short duration RCT including smoking asthmatics, combination salmeterol/fluticasone proprionate has been shown to improve lung function and to reduce bronchial hyperresponsiveness to methacholine compared to fluticasone alone.36 A recent prospective observational study was carried out to explore the real-life effectiveness of extra-fine HFA BDP in combination with the long-acting beta-agonist therapy, formoterol (FOR) in smokers with moderate-to-severe persistent asthma.37 The one-year outcomes were improvement in lung function (measured as improvement FEV1) and improvement in asthma control, evaluated using the 6-question Asthma Control Questionnaire (ACQ6) and Global Initiative for Asthma (GINA). The effectiveness cohort comprised 568 asthma patients (123 smokers). Treatment with extra-fine HFA BDP/FOR via metred-dose inhaler was associated with significant (P<0.0001) improvements in pulmonary function (+7.1% in FEV1 percent predicted), ACQ6 (-1.32) and GINA control (improvement of control in 49.8% of patients) irrespective of smoking status (current, former, or ex). The study authors concluded that extra-fine HFA BDP/FOR is an effective management option in adult asthma patients, including smokers, treated in routine care.
Based on the earlier-presented data on the potential role of LTRA therapy in smoking asthma (and given their different mechanisms of action), another potentially useful treatment option for smoking asthmatics could be extra-fine ICS in combination with LTRA therapy. However further data, from both real-life effectiveness studies and efficacy trials, are required.
Alternative study designs
The association between cigarette smoking and diminished ICS efficacy may be unequivocal and the rationale for excluding current smokers from asthma efficacy trials may be sound, but their exclusion has led to gaps and bias in the evidence base. If all patients who are unlikely to respond to a therapy are excluded from an efficacy trial, the result is an enriched population of likely-responders and an inflated expectation of the potential effect size achievable when the therapy is prescribed at population level. For clinicians treating asthma patients with known smoking habits, there remains the challenge of making prescribing decisions for that patient without the benefit of a strong evidence base for the likely effectiveness of the treatment options available.
In order to address the limitations of the RCT evidence base in this area, it is valuable to draw on (and to conduct) supplementary studies of different, complementary design. Pragmatic trials and observational studies (so-called "real-life studies") are often designed with much broader inclusion and exclusion criteria (and less rigorous levels of patient-clinician interaction) than is typical of RCTs.
Although such studies may lack the internal validity achievable in RCTs, they are valuable in the generalizablity of their findings and their ability to evaluate outcomes in patient subgroups of particular note, as illustrated in the aforementioned studies of extra-fine ICS (monotherapy and in combination with long-acting bronchodilator therapy) in smoking asthma. They can be used to assess clinical hypotheses and to guide and inform the design of RCTs to test those hypotheses in a more rigorous setting.
The value and utility of such studies is gaining increasing recognition among clinicians, so much so that leading respiratory experts have come together in 2013 to form The Respiratory Effectiveness Group (www.effectivenessevaluation.org) a not-for-profit, independent, collaborative network designed to drive forward the field of real-life respiratory research over the next 5 years, advocating use of observational studies and pragmatic trials to complement RCTs, standardizing analysis methods and setting quality benchmarks to help improve the quality as well as the profile of the field.
A focus of REG activities will be conducting high-quality observational studies and pragmatic trials that will help to "plug the gaps" in the RCT evidence base, particularly around the effectiveness of interventions in patient groups typically excluded from RCTs. Indeed, a recent REG-funded observational database study explored differences in reliever therapy dependence in asthma patients across different smoking categories. A current smoking status was associated with significantly higher mean daily short-acting bronchodilator usage compared to that of exsmokers (P<0.001) and non-smokers (P<0.001), suggesting to the presence of more severe asthma symptoms and greater dependence on rescue medication among smokers.38
CONCLUSIONS
RCTs are crucial in establishing the efficacy of new interventions and are a cornerstone of the drug licensing process, but they are limited in the extent to which their findings can be extrapolated to the routine care environment and to the diversity of patients treated in everyday practice. Pragmatic trials and observational studies can provide complementary data to help inform decisions about appropriate treatment strategies in subgroups of patients of particular clinical interest.
Cigarette smoking is known to diminish the efficacy of traditional ICS therapies and smoking asthma is increasingly being regarded as an asthma phenotype that requires recognition and consideration when making prescribing decisions. Drawing on the evidence available, asthma treatment options for smokers who refuse to quit may include: higher-dose prescribing (or dose escalation) of traditional ICS and/or use of extra-fine ICS formulations to target the inflammation present in the small airways; use of LTRA therapy as an alternative to traditional ICS therapy, and possibly also extra-fine ICS in combination with long-acting beta-agonist or with LTRA therapy. However, more studies in all of these areas are required.
A diversity of different approaches and study designs should be used to further evaluate potential treatment options for smoking asthma - observational studies to test hypotheses and to inform RCTs and pragmatic trials, and also where traditional RCTs are not feasible due to budget, time and/or ethical considerations. Only by integrating different elements of the evidence jigsaw can a fuller appreciation of the clinical picture be achieved and used to guide inform timely decision making.


 XML Download
XML Download