

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The bronchial epithelium has traditionally been recognized as a physical barrier protecting the host from its environment. However, epithelial cells play a central role in the regulation of airway immunity, affecting inflammation and host defenses in diseases of the airway. Epithelial cells release a wide range of proinflammatory mediators and multifunctional cytokines in response to exposure to inhaled environmental factors or microorganisms. The precise mechanisms are not fully understood, but epithelial cells are thought to play a major part in the regulation of host inflammatory status as well as airway structure and function.1,2
Lipid rafts are subdomains of the epithelial cell membrane that contain high concentrations of cholesterol and glycosphingolipids. They interact with one another and pack tightly together to form cell membrane structures. Thus, lipid rafts provide a platform for multiple signaling pathways and act as key modulators of certain disease pathways.3 Lipid entities render lipid rafts insoluble in nonionic detergents and cause them to separate from their surroundings. Cholesterol, the most abundant lipid component of animal cell membranes, regulates membrane fluidity and plays a crucial role in the formation and stabilization of membrane microdomains. It is also an important contributor to cell-cell adhesion, migration, and even endocytosis.4-7 However, despite increasing interest in the bronchial epithelium, the possible role of cholesterol in inflammation of the airway or the development of asthma has not been investigated.
Among the numerous cytokines and chemokines released from human airways, interleukin-8 (IL-8) is a representative chemokine expressed by bronchial epithelial cells. IL-8 mediates cell migration during inflammation of the airway.8,9 In addition, patients with severe asthma have increased levels of IL-8 in their BAL fluids. In addition, various stimuli, including house dust mites, cockroaches, and microbes, induce IL-8 production in bronchial epithelial cells and promote inflammation.10-13
Here, we investigated the effect of cholesterol depletion in airway epithelial cells on the production of IL-8 and its association with inflammation of the airway.
MATERIALS AND METHODS
Cell culture
The human epithelial-like lung carcinoma cell line A 549 was obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in F12K medium (Sigma, St. Louis, MO, USA) supplemented with 10% fetal bovine serum containing 100 U/mL penicillin and streptomycin (GibcoBRL, Grand Island, NY, USA). At all stages of culture, the cells were maintained in an incubator at 37℃ with 5% CO2.
Cholesterol depletion and repletion
Methyl-β-cyclodextrin (MβCD; Sigma) binds specifically to cholesterol to disturb the association of proteins with lipid rafts.14 It is therefore presumed to change the structure and function of the cell membrane by disrupting lipid rafts.15-17 A stock solution of 10% MβCD in phosphate-buffered saline (PBS) was stored at 4℃. This solution was used at concentrations of 0.5, 1, and 2% (v/v). After serum starvation for 24 h, cells were incubated with the indicated concentrations of MβCD for 1 h at 37℃ for cholesterol depletion. The culture medium was replaced with fresh serum-starved medium at the indicated times, and the cells were maintained at 37℃ in an incubator with 5% CO2. For cholesterol repletion, MβCD-treated cells were incubated for 1 h in the presence of 70 µg/mL cholesterol and 0.2% MβCD. The cells were then further incubated in fresh serum-free medium in an incubator.
Cell viability
A 549 cell viability at various concentrations of MβCD was measured with a Cell Counting Kit-8 (Dojindo, Kumamoto, Japan). The day before the experiment, 100 µL cells were seeded into 96-well microplates at a density of 1×104 cells per well. After 24 h of incubation, 10 µL cells per well were treated with various concentrations of MβCD for 1 h, followed by incubation with an additional 10 µL Cell Counting Kit-8 solution for 1 h. The absorbance was then measured at 450 nm with an enzyme-linked immunosorbent assay (ELISA) reader; the compensated absorbance was 590 nm.
Lipid extraction and cholesterol assay
A 549 cells pretreated with MβCD to deplete cholesterol were washed in cold PBS buffer and extracted with Folch solution containing chloroform and methanol at a 2:1 ratio. The cholesterol released by the Folch method was solubilized in the organic phase, and the amount of cholesterol was measured with a cholesterol probe following vaporization of the organic phase. The organic phase was dried under a speed vacuum (Savant Instruments Inc., New York, NY, USA) for 30 min, then solubilized with 2-propanol and 10% Triton X-100. Membrane cholesterol was assayed using a Cholesterol/Cholesteryl Ester Quantitation Kit (Calbiochem-Novabiochem, La Jolla, CA, USA), according to the manufacturer's instructions, Samples and standards were loaded and reacted by the addition of a reaction mixture containing cholesterol buffer, probe, and enzyme without light for 1 h. The absorbance at 570 nm was measured with an ELISA reader; the compensated absorbance was 590 nm. A BCA Protein Assay Kit (Pierce, Rockford, IL, USA) was used to quantify the extracted proteins, which were collected with Mammalian Protein Extraction Reagent (Pierce) and normalized to the volume.
Measurement of human IL-8 and MAP kinase (MAPK) assay
After treatment with 0.5% MβCD for 1 h, cells were treated with fresh serum-free medium and incubated for 2, 5, 10, and 24 h at 37℃. They were then stored at -70℃ until they were assayed. The IL-8 levels in the culture supernatants were determined using a specific ELISA against human IL-8 (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions. To investigate the effect of changes in transcription factor activity on IL-8 expression in human lung epithelial cells, the selective MAP/extracellular signal-regulated kinase (ERK) [MEK] inhibitor PD98059 or U0126, c-JUN NH2-terminal kinase (JNK) inhibitor JNK II Inhibitor, or p38 MAPK inhibitor SB203580 or SB202190 (Calbiochem-Novabiochem) was added 1 h before stimulation with MβCD and was present throughout the incubation period.
RNA preparation and real-time polymerase chain reaction (PCR)
Reverse transcription-PCR (RT-PCR) was used to examine the mRNA expression of IL-6, IL-8, and TNF-α. Total RNA was isolated from cells using Trizol reagent (Invitrogen, Carlsbad, CA, USA) prior to cDNA synthesis. PCR amplification was carried out using specific primer pairs. The primer sequences used were obtained from Primer Bank (http://pga.mgh.harvard.edu/primerbank). Fold changes were calculated using the comparative ΔΔCt method. The gene encoding glyceraldehyde-3-phosphate dehydrogenase (GADPH) was analyzed as an internal control in each sample.
Western blot analysis
MβCD-treated cells were lysed with lysis buffer (50 mM HEPES, pH 7.5; 150 mM NaCl; 1 mM EDTA; 10% glycerol; and 0.5% NP-40). Protein samples were mixed with 5× SDS-PAGE buffer containing β-mercaptoethanol and heated at 95℃ for 5 min. Wholecell lysates (30 µg) were separated by 10% SDS-PAGE, then electrotransferred to nitrocellulose membranes (GE Healthcare, Stockholm, Sweden). The membranes were blocked for 1 h with 5% skim milk in TBST buffer (10 mM Tris-HCl, pH 7.5; 150 mM NaCl; and 0.2% Tween 20), then washed three times with TBST at room temperature. The blots were incubated overnight with specific antibodies (1:1,000) in 5% skim milk in TBST buffer at 4℃. After incubation with secondary antibodies (1:2,000), the signal was detected using the ECL Plus (GE Healthcare) and CLXPosure™ film (Thermo Scientific, Waltham, MA, USA) or an ImageQuant™ LAS 4,000 Mini Biomolecular Imager (GE Healthcare).
RESULTS
Induction of cholesterol depletion by MβCD in A 549 cells
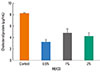
We estimated the amount of cholesterol in the growth media of control and MβCD-treated cultures. After treatment with 0.5, 1, or 2% MβCD for 1 h at 37℃, the cells showed a decreased amount of cholesterol (3.15±0.52, 4.73±0.75, and 4.17±0.68 µg/mL, respectively) compared to untreated cells (8.22±0.11 µg/mL). However, there was no significant difference in the amount of cholesterol among the MβCD-treated groups. In addition, the amount of cholesterol in the cell was at its lowest following treatment with 0.5% MβCD, showing an approximately 60% decrease compared to the control (Fig. 1). Subsequent experiments were thus carried out using epithelial cells treated with 0.5% MβCD.
Alteration of cell viability by MβCD
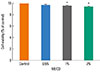
To determine whether cholesterol depletion by MβCD had any effect on cell viability, we compared the untreated cells to the A 549 cell cultures treated with 0.5, 1, or 2% of MβCD for 1 h. There were no dramatic changes in overall cell viability compared to the control (96.8, 95.3, and 94.2%, respectively, Fig. 2) but statistical differences showed in 1 and 2% of MβCD-treated cells. We concluded that 0.5% MβCD should be used in our subsequent experiments.
Effect of cholesterol depletion on human IL-8 production
The effect of cholesterol depletion on IL-8 production was examined in airway epithelial cells treated with 0.5% MβCD. Cholesterol-depleted cells produced more IL-8 than control cells at different time points (2, 5, 10, and 24 h). IL-8 production increased in a time-dependent manner, with maximum production at 24 h. Cells treated with 0.5% MβCD produced 300.4±7.9 pg/mL at 2 h, 477.7±1.5 pg/mL at 5 h, 816.7±55.2 pg/mL at 10 h, and 867.6±38.9 pg/mL at 24 h compared to 40.8±2.3 pg/mL at 2 h, 80.4±0.2 pg/mL at 5 h, 158.2±8.3 pg/mL at 10 h, and 244.6±6.0 pg/mL at 24 h for control cells. We verified that the IL-8 level decreased after treatment with both 0.5% MβCD and 70 µg/mL cholesterol, similar to in the control cells (Fig. 3A). When tested by RT-PCR with GAPDH as an internal control, the A 549 cells depleted of cholesterol with 0.5% MβCD showed greater IL-8 mRNA expression. IL-8 expression peaked at 2 h and returned to baseline 24 h after MβCD stimulation (Fig. 3B).
Role of MAPK signaling in IL-8 production
To investigate the link between IL-8 production in cholesterol-depleted cells and the MAPK signaling pathway, which is involved in airway inflammatory diseases, we examined the effect of MAPK inhibitors on IL-8 production. IL-8 production was decreased by pretreatment with the ERK inhibitor U0126 (50 µM), but JNK inhibitor II (100 µM) and the p38 MAPK inhibitor SB202190 (50 µM) did not significantly reduce IL-8 production (Fig. 4A). The level of IL-8 was decreased by almost 80% by U0126 but by only 10.9 and 31.4% by JNK inhibitor II and SB202190, respectively, compared to MβCD treatment alone.
To demonstrate the intracellular activation of MAPK signaling, we examined the phosphorylation of ERK, p38 MAPK, and JNK in MβCD-treated cells from 0 to 60 min. As shown in Fig. 4B, we detected small amounts of phosphorylated ERK 1/2 MAPK in untreated A 549 cells, whereas treatment with MβCD induced ERK 1/2 MAPK phosphorylation. ERK 1/2 MAPK activation was maximized at 30 min, but neither p38 MAPK nor JNK activation was detected.
Effect of cholesterol depletion on the production of other cytokines
To identify the effect of cholesterol depletion on inflammatory responses in the airway, we examined the production of other cytokines released from A 549 cells. When tested by RT-PCR with GAPDH as an internal control, cholesterol depletion by MβCD resulted in elevated IL-6 and TNF-α mRNA expression in the A 549 cells. IL-6 and TNF-α expression peaked 2 h after MβCD stimulation (Fig. 5A and B).
DISCUSSION
The epithelium, which represents the first contact surface with the external environment, forms a tight and impermeable barrier for the maintenance of tissue homeostasis under normal circumstances.1 The structural integrity of the epithelium is also maintained through cell-cell and cell-extracellular matrix interactions. However, air pollutants or infectious particles can increase the permeability of the epithelium and penetrate the airway, inducing epithelial cell damage or death. Allergens such as house dust mites and pollen can also disrupt the integrity of the mucosa through the direct extracellular cleavage of tight junctions.2 Such allergens also enhance inflammatory pathways that make the epithelium interact with other cells.18,19 In addition, epithelial cells can recruit and modulate immune cells to cause persistent inflammation through adhesive interactions, selective cytokine release, and cell-specific receptor expression. Thus, epithelial cells can modulate the structure and function of the airway by contributing to host defenses and innate immunity.1,2
A recent innovation in our understanding of the role of epithelial cells in inflammation of the airway has to do with the influence of impaired innate immunity and epithelial cell injury on the progression of inflammation, especially in the case of asthma.18-20 The airway epithelium in asthma patients is damaged, with the disruption of tight junctions accompanied by a loss of junctional proteins in response to environmental factors such as dust mite allergens, fungal allergens, viral infections, and smoke.21,22 The damaged epithelium results in barrier dysfunction, epithelial fragility, and abnormal repair responses in the asthmatic airway, creating a condition that is similar to inflammatory conditions of the skin such as atopic dermatitis or those of the gut in the case of food allergy.18,19 Many studies have focused on compromised tight junctions and epithelial cell protein complexes to determine the relationship between asthma and the airway epithelium.18-22 However, the role of lipid components of the epithelial cell membrane in inflammation of the airway has not been comprehensively studied.
Lipid rafts are dynamic nanoscale membrane microdomains that contain large amounts of cholesterol and sphingolipids involved in signaling platforms.3 Cholesterol, an essential component of the plasma membrane, has been implicated in the regulation of membrane fluidity, permeability, and cell surface receptor function.4-6 Because cholesterol contributes to the establishment of a cornified barrier, it plays a crucial role in keratinocytes. Atopic dermatitis has even been shown to occur in parallel with keratinocytes disrupted by lipid rafts.17 We hypothesize that inflammatory diseases of the airway are influenced by airway epithelial cells with disrupted lipid rafts, similar to keratinocytes in atopic dermatitis.
To clarify the relationship between cholesterol and inflammation of the airway, we used MβCD to damage lipid rafts in airway epithelial cells. We identified a concentration of MβCD capable of inducing significant changes in cholesterol levels in the cell. We then demonstrated that the amount of cholesterol in the cell was significantly decreased after incubation with 0.5% MβCD, without effect on cell viability.
IL-8 is a major chemokine of the CXC chemokine subfamily that acts as a chemoattractant for neutrophils released from the bronchial epithelium.8 IL-8 plays a key role in triggering and perpetuating inflammation in patients with severe asthma. In addition, the expression and release of IL-8 increase in response to other stimuli such as allergens and infectious particles.10-13 Hence, we used IL-8 as an inflammatory marker in this study, and IL-8 production rose in cholesterol-depleted epithelial cells. Based on these results, we recognize that IL-8 production in human respiratory epithelial cells could be affected by the cholesterol content in the membrane through the depletion of cholesterol in the cell in the absence of any other allergen.
MAPK regulates IL-8 expression and release in response to various allergens and viruses in airway epithelial cells, resulting in an influence on cell signaling pathways in diseases of the airway. Among ERK, JNK, and p38 MAPK, ERK-regulated IL-8 production has been suggested as a distinct signaling process in chronic diseases of the airway.10-13 We investigated the effect of MAPK inhibitors on IL-8 production using U0126, JNK inhibitor II, and SB202190 and sought to determine whether IL-8 production in cholesterol-depleted cells was affected by the MAPK signaling pathway. Our results suggest that activation of the ERK pathway is a relatively strong contributor to IL-8 production in cholesterol-depleted cells, in agreement with previous studies of allergen-induced inflammation of the airway.10-13
In addition, we investigated the expression of other cytokines generated by airway epithelial cells. IL-6 can stimulate T cells and contribute to the development of Th2-mediated inflammation. It was previously demonstrated that epithelial IL-6 expression increased after exposure to fungi, house dust mites, or common respiratory viruses.1 In addition, airway epithelial cells produce pleiotropic cytokines, including TNF-α, which plays an important role in severe refractory asthma. Targeting TNF-α may be a novel therapeutic approach to treating asthma.23 We found that the mRNA expression of IL-6 and TNF-α was increased in cholesterol-depleted epithelial cells. Cholesterol depletion in the airway epithelium may be related to inflammatory responses through the production of several inflammatory products.
Genome expression profiling has been performed in cholesterol-depleted keratinocytes from patients with atopic dermatitis.17 What is interesting is that patients with genetic differences tend to be susceptible to both this condition and other related allergic diseases.24,25 Such large-scale genome profiling and genetic experiments have confirmed that the disruption of lipid rafts can lead to inflammatory disease under certain circumstances.
In conclusion, our findings suggest that the disruption of lipid rafts plays an important role in inflammation of the airway. Bronchial epithelial cells with cholesterol depletion released a large amount of IL-8, and IL-8 production was regulated by the MAPK signaling pathway, predominantly through ERK. In addition, cholesterol depletion in airway epithelial cells induced the elevation of other proinflammatory cytokines generated by the airway epithelium. Considering that the bronchial epithelium may be a novel target for new therapies for inflammatory diseases of the airway, additional studies of cholesterol and lipid rafts in human airway epithelial cells are needed.



 XML Download
XML Download