

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Nasal polyposis is a chronic inflammatory disease of the sinonasal mucosa that involves pathognomonic infiltration of inflammatory cells, particularly eosinophils, and tissue remodeling, such as basement membrane thickening, extracellular matrix accumulation, and fibrosis. Angiogenesis and microvascular remodeling are elements of tissue remodeling in nasal polyposis.1 Angiogenesis depends on a coordinated balance of the levels of vascular endothelial growth factor (VEGF),2 a 45-kDa, heparin-binding homodimeric glycoprotein. VEGF may improve microvascular permeability and therefore contribute to the edema frequently observed in nasal polyps. Moreover, VEGF, by increasing microvascular permeability, allows the extravasation of plasma proteins that participate in the accumulation of the extracellular matrix, thereby accelerating nasal polyp growth.1
Among cyclooxygenase-derived prostanoids, prostaglandin E2 (PGE2) plays both pro- and anti-inflammatory roles in airway inflammation.3 PGE2 was reported to potently inhibit fibroblast-mediated tissue repair, including chemotactic recruitment, proliferation, production, and extracellular matrix remodeling.4 Prostaglandins, particularly PGE2, play the principal role in modulating the inflammatory process in patients with chronic nasal polyposis.3 Although aspirin-intolerant asthmatic patients with nasal polyposis have been reported to have a low level of PGE2, which protects the airways against inflammation and promotes normal airway function,5 some investigations have suggested that PGE2 production, which can have pro-inflammatory effects, contributes to the development of nasal polyposis.5,6 While PGE2 induces VEGF production in rheumatoid arthritis synovial fibroblasts and human prostate cancer cells, it reduces tumor necrosis factor-α production in rodent macrophages and macrophage-colony-stimulating factor production in human monocytes.7 PGE2 signals through four G-protein coupled receptors [E-prostanoid (EP) receptors 1-4].8
The ability of PGE2 to induce or suppress various pathways involved in the inflammatory process is an indication of the complex activities of its receptors.9 The EP1 receptor is related to phospholipase C and phosphoinositol turnover and stimulates the release of intracellular calcium. EP2 and EP4 receptors increase cyclic adenosine monophosphate (cAMP) levels by activating adenylate cyclase, whereas the EP3 receptor mediates multiple signal pathways, including inhibition or stimulation of cAMP levels, activation of phospholipase C, and mobilization of intracellular calcium. Hence, the role of PGE2 in inflammatory and immune responses is complicated; it is temporally defined and largely influenced by EP receptor expression. More importantly, several reports have shown that specific EP receptors play a major role in regulation of immune responses and airway inflammation other than nasal polyp-derived fibroblasts (NPDFs).8,10 NPDFs are key cells in nasal polyp architecture. They are stimulated by pro-inflammatory cytokines and eosinophil-secreted growth factors. Stimulated fibroblasts contribute to the inflammatory process by releasing inflammatory mediators.1 The predominance of a particular EP receptor type may determine the mechanism of PGE2-mediated action in a particular cell type. PGE2 stimulates VEGF production in rat gastric fibroblasts through the EP4 receptor and in human synovial fibroblasts through the EP2 and EP4 receptors.10,11 However, it remains to be discovered whether PGE2 participates in producing soluble mediators such as VEGF through specific EP receptors in NPDFs. To our knowledge, few in vitro studies have investigated the stimulatory effect of PGE2 on VEGF production in NPDFs and the relationship with specific EP receptor subtypes and PGE2-induced VEGF upregulation.
Therefore, in the present study, we assessed the stimulatory effect of PGE2 on VEGF production in NPDFs and further investigated whether specific EP receptor subtypes and signal transduction pathways are associated with PGE2-induced VEGF upregulation.
MATERIALS AND METHODS
Study subjects and design
Fibroblasts were obtained from eight patients (four females and four males; mean age, 32.3±5.2 years) who underwent endoscopic sinus surgery for nasal polyposis at the Department of Otorhinolaryngology, Soonchunhyang University College of Medicine. None of the patients were smokers, had a history of nasal allergy, asthma, or aspirin hypersensitivity, or had been treated with oral or topical anti-allergic agents during the previous 8 weeks. Written informed consent was obtained from all patients prior to surgery. The present study was approved by the Institutional Review Board of Soonchunhyang University College of Medicine.
After cell culture, we utilized reverse transcription-polymerase chain reaction (RT-PCR) to assess mRNA levels of various EP receptors in NPDFs, and then confirmed whether PGE2 increased VEGF mRNA and protein levels in a concentration and time-dependent manner using RT-PCR and enzyme-linked immunosorbent assay (ELISA) separately. To determine the type of EP receptor involved in VEGF production in NPDFs, various EP receptor agonists and antagonists were used and their effect evaluated by ELISA and immunofluorescence staining. In addition, we examined the effect of specific mediators of the cAMP-dependent signal transduction pathway on VEGF production by ELISA.
Reagents
The phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002 and cAMP activator forskolin were purchased from Sigma (St. Louis, MO, USA). Dulbecco's modified Eagle's medium (DMEM) was obtained from Invitrogen (Carlsbad, CA, USA). PGE2 was dissolved in DMEM with 10% heat-inactivated fetal calf serum, and then diluted to the desired concentration in complete medium for use in the experiments. PGE2, the protein kinase A (PKA) inhibitor KT5720, the EP1/3 receptor agonist sulprostone, the EP2 receptor agonist butaprost, the EP4 receptor agonist CAY 10580, the EP1 receptor antagonist SC51322, the EP2 receptor antagonist AH6809, the EP3 receptor antagonist L798106, and the EP4 receptor antagonist AH23848 were obtained from Cayman Chemical (Ann Arbor, MI, USA). Anti-human VEGF antibody was obtained from BD Biosciences (Minneapolis, MN, USA). The Quantikine human VEGF ELISA kit was purchased from R&D Systems (Minneapolis, MN, USA). The cAMP enzyme immunoassay kit was purchased from Assay Design (Ann Arbor, MI, USA).
Isolation and induction of NPDF
Nasal polyp tissues were cut into 2-3-mm3 pieces under sterile conditions. NPDFs were isolated from surgical tissues by enzymatic digestion with collagenase (500 U/mL; Sigma), hyaluronidase (30 U/mL; Sigma), and DNase (10 U/mL; Sigma). Briefly, following 2 hours of incubation in a culture plate in a 5% CO2 atmosphere at 37℃, cells were collected by centrifugation, washed twice, and resuspended in DMEM containing 10% (v/v) heat-inactivated fetal bovine serum and antibiotics: 2-glutamate (Invitrogen), 100 µg/mL penicillin, and 100 µg/mL streptomycin (Invitrogen). Cells were allowed to attach to the culture plate for 4 days. Nonadherent cells were removed by changing the medium. Fibroblasts were detached with EDTA solution (Invitrogen). After washing, cells were resuspended in medium and used for subsequent experiments. The fibroblast purity was >99% and was used for NPDFs. Cells were used at passage 4.
RT-PCR
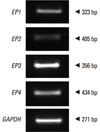
Reverse transcription was performed with 2 µg of RNA from each NPDF sample. Total RNA was denatured at 65℃ for 5 minutes. The total RNA concentration was determined by spectrophotometry. After cooling on ice, the following components were added to the samples: 5× reverse transcriptase buffer, 2.5 mM dNTPs, RNase inhibitor, and Moloney Murine Leukemia Virus reverse transcriptase. After 60 minutes at 37℃, the reverse transcriptase was inactivated by heating the mixture at 95℃ for 5 minutes. PCR was performed using primers for EP1 (sense sequence: 5'-GAT GGT GGG CCA GCT TGTC-3'; anti-sense sequence: 5'-GCC ACC AAC ACC AGC ATTG-3'; 323 bp), EP2 (sense sequence: 5'-GAC CGC TTA CCT GCA GCT GTAC-3'; anti-sense sequence: 5'-TGA AGT TGC AGG CGA GCA-3'; 405 bp), EP3 (sense sequence: 5'-AAG GCC ACG GCA RCT CAGT-3'; anti-sense sequence, 5'-TGA TCC CAT AAG CTG AAT GG-3'; 256 bp), EP4 (sense sequence: 5'-ACG CCG CCT ACT CCT ACA TG-3'; anti-sense sequence: 5'-AGA GGA CGG TGG CGA GAAT-3'; 434 bp), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (sense sequence: 5'-GTG GAT ATT GTT GCC ATC AAT GAC C-3', anti-sense sequence 5'-GCC CCA GCC TTC TTC ATG GTG GT-3'; 271 bp) supplied by Bioneer (Daejeon, Korea). PCR products were subjected to electrophoresis on a 2% agarose gel, stained with ethidium bromide, and visualized by ultraviolet fluorescence. To analyze the RT-PCR results semi-quantitatively, the gel images were scanned, and the intensities of the PCR products were measured using the ImageJ software (National Institutes of Health, Bethesda, MD, USA). The relative intensities of individual bands in a gel image were determined as ratios to the GAPDH band intensity. No PCR product was amplified in the negative reverse transcription reaction.
Immunofluorescence staining of VEGF protein
Cells were fixed in phosphate-buffered saline (PBS) containing 4% paraformaldehyde for 30 minutes, blocked with 3% bovine serum albumin, and incubated with monoclonal anti-VEGF (1:200 dilution) for 3 hours, and washed three times with PBS for 5 minutes. Cells were then incubated in goat anti-mouse IgG Alexa Fluor 488 (Invitrogen) at 1:100 for 1 hour, and then mounted on Vectashield from Vector Laboratories (South San Francisco, CA, USA) with 4',6-diamidino-2-phenylindole. Each stained tissue was captured and visualized using confocal z-stack laser scanning microscopy (LSM 700; Zeiss, Oberkochen, Germany).
Measurement of VEGF protein and cAMP
Cells were seeded at 2×105/well in six-well plates and allowed to attach for 24 hours. The medium was removed, cells were washed twice with PBS, and cultures were incubated in serum-free medium for 16 hours prior to treatment. Cells were treated with PGE2 and/or sulprostone, butaprost, and CAY10580 in serum-free medium containing 1% bovine serum albumin for 24 hours. PGE2, sulprostone, butaprost, and CAY10580 were dissolved in absolute ethanol. To examine VEGF induction by forskolin, cells were treated with forskolin for 24 hours. For inhibitor studies, cells were pretreated with the respective inhibitors at the indicated working concentrations for 1 hour in serum-free medium prior to addition of PGE2. SC51322, AH 6809, L-798106, AH 23848, KT5720, and LY294002 were solubilized in DMEM. After treatment, the medium was collected and VEGF levels therein measured using the Quantikine human VEGF ELISA kit (R&D Systems). Cells were washed with PBS and cell lysed with radioimmunoprecipitation assay buffer. Protein concentrations media and cell lysates determined by the bicinchoninic acid method according to the manufacturer's instructions (Pierce; Rockford, IL, USA).
To assay cAMP, cells were seeded in six-well plates as described above. The doses of PGE2, CAY10580, AH23848, forskolin, KT5720, and LY294002 for the VEGF assay were used in serum-free medium, and cells were collected after 30 minutes. After treatment, cells were washed once with PBS before lysing with 0.5 mL of 0.1 M HCl and Triton X-100 (0.1%) for 15 minutes. Cell lysates were subsequently collected and subjected to the cAMP enzyme immunoassay according to the manufacturer's instructions (Assay Design, Ann Arbor, MI, USA). Protein concentrations in cell lysates were determined as above. For all experiments, the amounts of solvent were normalized among the treatments and controls.
Statistical analysis
All experiments were repeated at least three times. Comparisons among multiple groups were performed by one-way analysis of variance with Tukey's post hoc comparisons. A P value less than 0.05 was deemed to indicate statistical significance. All statistical outcomes based on the two-sided test were completed using the SPSS software (version 12, SPSS Inc., Chicago, IL, USA).
RESULTS
Expression of EP receptors in NPDFs
To determine EP receptor expression, RT-PCR was performed for EP1, EP2, EP3, EP4, and GAPDH mRNA. All PCR products could be separated by size. GAPDH was used as a positive control for RNA integrity in the RT-PCR procedure (Fig. 1).
Effect of PGE2 on VEGF production
To determine whether PGE2 regulated VEGF production, RT-PCR and ELISA were used. PGE2 significantly increased VEGF mRNA expression in a concentration and time-dependent manner (Fig. 2A and C). After 2 days, VEGF production of cultures stimulated with PGE2 increased significantly in a concentration and time-dependent manner (Fig. 2B and D).
Effect of EP receptor-specific agonists and antagonists on VEGF production
To evaluate the effect of the EP receptor on VEGF production, we treated fibroblasts with EP receptor-specific agonists and quantified VEGF production by ELISA. CAY 10580 (10 µM) significantly increased VEGF production in contrast to sulprostone (10 µM) and butaprost (10 µM) (Fig. 3A). We confirmed these results using receptor-specific antagonists to inhibit the effect of PGE2. The various EP receptor antagonists (10 µM) were added to the medium 1 hour before addition of PGE2 (20 µM), and VEGF production was determined by ELISA. EP receptor antagonists other than AH 23848 showed little effect on VEGF release from PGE2-stimulated fibroblasts. Only AH 23848 significantly decreased VEGF production (Fig. 3B).
To investigate whether the PGE2-induced VEGF production in NPDFs was regulated by the EP4 receptor, immunofluorescence staining for VEGF expression was performed. NPDFs treated with PGE2 alone showed strong VEGF immunoreactivity. Control and NPDFs treated with PGE2 and an EP4 receptor antagonist showed only blue-stained nuclei. By contrast, NPDFs treated with PGE2 and NPDFs treated with EP4 receptor agonist showed blue-stained nuclei with a greenish cytoplasm (Fig. 3C).
Effect of cAMP on VEGF production through EP4 signaling
cAMP production was evaluated following treatment with various PGE2 concentrations. cAMP production was increased by PGE2 in a concentration-dependent manner (Fig. 4A). Forskolin (40 µM) was utilized to determine whether cAMP was involved in VEGF production. Forskolin stimulated significant VEGF production (Fig. 4B). To determine whether cAMP affects VEGF production through EP4 signaling, NPDFs were treated with PGE2, CAY 10580 and AH 23848. PGE2 and CAY 10580 significantly increased cAMP level compared to control. When AH 23848 was added to the medium before the treatment of PGE2, cAMP level was significantly decreased (Fig. 4C).
Effect of PKA and PI3K on VEGF and cAMP production
We next determined whether cAMP-dependent PKA and/or PI3K mediated the effect of PGE2 in stimulating VEGF production. KT5720 (1 µM) or LY294002 (30 µM) was added 1 hour before PGE2 treatment (20 µM). Both KT5720 and LY294002 decreased VEGF production significantly (Fig. 5A). To elucidate the signal transduction pathway leading to VEGF production, we compared cAMP release by cells treated with PGE2 alone, PGE2 and KT5720, or PGE2 and LY294002. KT5720 significantly inhibited cAMP release, but LY294002 did not (Fig. 5B).
DISCUSSION
We aimed to determine whether PGE2 regulated VEGF production by NPDFs and whether EP receptors mediated this effect. ELISA and RT-PCR showed an increase in VEGF mRNA and protein in NPDFs, which was dependent on the PGE2 concentration and treatment time. These results indicate that PGE2 is a stimulator of VEGF production in NPDFs. Moreover, the EP4 receptor likely plays a major role in mediating VEGF production in NPDFs. Immunofluorescence staining for VEGF release in cells treated with an EP4 receptor agonist or antagonist supported these results.
cAMP can activate the classic PKA pathway, which stimulates VEGF production in response to prostacyclin.12 Recent studies have shown the involvement of cAMP-dependent signal transduction pathways, such as the PKA and PI3K pathways through binding sites on the VEGF promoter, to be responsible for VEGF induction via the EP4 receptor.13,14 The available data suggest that the EP4 receptor preferentially couples to the G-protein that increases cAMP concentrations and also activates the PI3K pathway.15 Our study showed that cAMP release depended on the PGE2 concentration and stimulated VEGF production in NPDFs. Additionally, both KT5720, a PKA inhibitor, and LY294002, a PI3K inhibitor, significantly antagonized PGE2 stimulation of VEGF production. LY294002 failed to block the PGE2-induced increase in cAMP, whereas KT5720 blocked the PGE2-induced increase in cAMP. These findings suggest that PGE2 stimulation of EP4 receptors result in the activation of cAMP/PKA signaling pathway and PI3K signaling pathway.
Although the etiology of nasal polyposis and the pathophysiological mechanisms leading to polyp formation are poorly understood, evidence suggests that damage to the mucosal epithelium is accompanied by extracellular matrix accumulation and inflammatory cell infiltration.16 NPDFs are key cells in the nasal polyp architecture and respond to the pro-inflammatory cytokines and growth factors secreted by eosinophils.16 Stimulated fibroblasts contribute to the inflammatory process by releasing inflammatory mediators.17,18 One characteristic of nasal polyps is substantial tissue edema. This edema has been attributed to local VEGF production, which plays an important role in angiogenesis and modulating capillary permeability.2
PGE2 has traditionally been identified as a prevalent inflammatory mediator in many tissues and inflammatory diseases, including chronic rhinosinusitis.19,20 PGE2 is produced by many cells, including fibroblasts, macrophages/monocytes, dendritic cells, and some malignant cells, and exerts its effects through G protein-coupled receptors. PGE2 increases proliferation, metastatic capacity, and production of pro-angiogenic factors.21 Recently, the EP receptor subtypes were cloned, and agonists specific for each were purified.22 EP2 and EP4 stimulate G proteins and mediate increases in cAMP, whereas EP3 decreases cAMP levels. The physiological functions of PGE2 are thought to be mediated through interactions with four distinct prostaglandin receptors associated with different signal transduction pathways.9,10 For example, PGE2 inhibited lung fibroblast chemotaxis through the cAMP-dependent PKA pathway.23 Accumulating evidence demonstrates that EP receptors (individual or in combination) can target various signaling molecules in many biologic models,24,25 including upregulating VEGF by PKA-dependent pathways and upregulating PI3K. Because of the complexity of PGE2 and the signaling system of its EP receptors, their effects in specific cell types should be determined.
Few reports have described the effect of PGE2 on VEGF production through the role of EP receptor isoforms in NPDFs. Our study is to our knowledge the first to show that PGE2 stimulates VEGF production through the EP4 receptor in NPDFs. Signaling through cAMP/PKA pathway and PI3K pathway appear to mediate this effect, at least part. By stimulating VEGF production, PGE2 may induce nasal remodeling while stimulating vascular responses. The present findings provide new insights into the mechanisms underlying the regulation of VEGF production in NPDFs, a function that is mediated by the EP4 receptor. Demonstration of a physiologic role for PGE2-driven VEGF production, the main factor of angiogenesis that is involved in the pathogenesis of nasal polyposis, however, will require in vivo research. Additionally, further studies are necessary to elucidate the relationship between the signal transduction pathway and PGE2-induced VEGF production in NPDFs.
In conclusion, our data suggest that PGE2 stimulates VEGF production via the EP4 receptor in NPDFs. Many questions remain regarding selective antagonists of the EP4 receptor that might be effective in preventing the development of nasal polyps.




 XML Download
XML Download