

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Asthma is defined by the Global Initiative for Asthma as "a chronic inflammatory disorder of the airways in which many cells and cellular elements play a role; the chronic inflammation is associated with airway hyperresponsiveness (AHR) that leads to recurrent episodes of wheezing, breathlessness, chest tightness and coughing, particularly at night or in the morning; these episodes are usually associated with widespread, but variable airflow obstruction within the lung that is often reversible either spontaneously or with treatment".1 Asthma was recognized in Ancient Egypt and was treated by drinking an incense mixture known as kyphi.2 Asthma was first named and recognized as a specific respiratory problem by Hippocrates, circa 450 BC. During the 1930-50s, asthma was considered to be a psychosomatic disorder, and its etiology was considered to be psychological, with treatment often based on psychoanalysis.3 In the 1980s, advances in medical research methodologies, such as studies on bronchoscopic biopsy and induced sputum in asthmatic patients, demonstrated that asthma is characterized by chronic inflammatory disease in the airways.4,5
The World Health Organization estimates that 300 million people worldwide currently suffer from asthma. Moreover, asthma is the most common chronic disease among children, leading to approximately 250,000 deaths per year.6-8 Although the prevalence of asthma is 7%-10% worldwide,9 there is great regional disparity in its prevalence globally, with higher asthma rates tending to exist in more developed and westernized countries than developing countries,10 with as much as 20- to 60-fold differences. Asthma is a heterogeneous inflammatory disease involving many cell types.11 Asthma is clinically classified according to the frequency of symptoms, forced expiratory volume in 1 second, and peak expiratory flow rate.12 Although asthma can be classified based on the severity of symptoms, there is no advanced classification method that allows differentiation of asthma subgroups for asthma pharmacotherapy.13 Therefore, identifying subgroups that respond well to different types of treatment is currently an important goal of asthma research.
Our understanding of the pathogenesis of asthma has changed dramatically. Whereas mast cells and eosinophils were initially believed to play a central role in driving the airway inflammation associated with asthma, new data imply that T helper cells are critical.14 In recent decades, immunologic mechanisms have been studied extensively, and asthma is now regarded as an IgE-mediated sensitization to inhaled allergens with a Th2 cell response and subsequent eosinophilic inflammation and AHR.15 The "Th2 asthma hypothesis" is based on the assumption that IgE and eosinophils play crucial roles in the pathogenesis of asthma; Th2 cytokines are believed to regulate IgE synthesis, and eosinophil numbers and activity are thought to play a major role in driving its pathogenesis.16,17 Additionally, it has been proposed that immune deviation toward a Th1 response can protect against asthma, since Th1 cells antagonize the functions of Th2 cells.18,19 However, much evidence using sputum induction and/or bronchoalveolar lavage (BAL) techniques to measure and characterize airway inflammation in asthma patients indicates that a substantial proportion of asthma cases have an underlying pathology that is clearly different from Th2 eosinophilic asthma.20-23 Specifically, such patients have severe and persistent asthma in the absence of eosinophilic inflammation, and may experience an exacerbation of asthma without an increase in eosinophilic inflammation.20 In addition, persistent asthma may be associated with the presence of significant neutrophilic inflammation,23 predominant neutrophilic inflammation has been linked to corticosteroid resistance in stable asthma patients,24 and neutrophils rather than eosinophils often predominate in patients with difficult-to-control asthma.21 In this review, we discuss recent progress in knowledge of the immunological pathogenesis of allergic asthma induced by inhaled allergens, based mainly on data from animal experiments.
Go to : 
ANIMAL MODELS OF ALLERGIC ASTHMA: GENERAL CONSIDERATIONS
Animal model studies form the basis for much of our current understanding of the pathophysiology of allergic asthma, and are central to the preclinical development of drug therapies. Early animal models of allergic asthma used a variety of species and focused on the AHR phenomenon.25 Rat models have been widely employed, notably the Brown-Norway strain because of its propensity to exhibit a later allergic response following antigen challenge.26 The monkey has attracted attention as a useful model due to its greater human relevance than rodent models.27 Nevertheless, most asthma-related research has been conducted using the mouse, which is currently the species of choice for asthma research involving animals.
In the early 1980s, a greater awareness of the role of inflammation in asthma was driven by an increased understanding of allergic responses and observations that human asthmatics frequently exhibited marked symptoms when challenged with antigens of various kinds. The first inflammatory cell to be linked firmly to asthma pathogenesis was the eosinophil,28,29 followed soon after by the T cell.30 Research focused on allergic inflammation by sensitizing and challenging animals with a variety of foreign proteins with or without adjuvants has led to an increased understanding of the immunological factors that mediate the inflammatory response and its physiological expression in the form of AHR. Allergic asthma mouse models are generated by sensitization to a foreign protein, most commonly ovalbumin (OVA). Sensitization is accomplished by injecting the protein intraperitoneally along with an adjuvant, typically aluminum hydroxide, which elicits an inflammatory reaction in the lungs that is characterized by an influx of eosinophils and AHR. Despite extensive use of this sensitization strategy, this model has two major drawbacks with respect to clinical relevance: the peritoneal sensitization of allergen may differ from an initial immune response to inhaled allergens, and the use of aluminum hydroxide induces eosinophilic airway inflammation, which reflects only a subset of eosinophilic asthma.
Go to :
ROLES OF HELPER T CELL SUBSETS IN ALLERGIC ASTHMA PATHOGENESIS
Results from animal model studies have implied that allergic asthma involves a complex interplay between the innate and adaptive immune systems. Adaptive immunity begins with naïve T cells that differentiate into T helper cells that potentially regulate the fate, function, and location of a variety of immune and inflammatory cells. Different subsets of T helper cells, such as Th1, Th17, and Th2 cells, have been defined on the basis of the cytokines they secrete. Th1 and Th17 cell production is promoted primarily by IL-12 and IL-6, respectively,31-33 whereas differentiation into Th2 cells occurs in the presence of IL-4 (Fig. 1).34
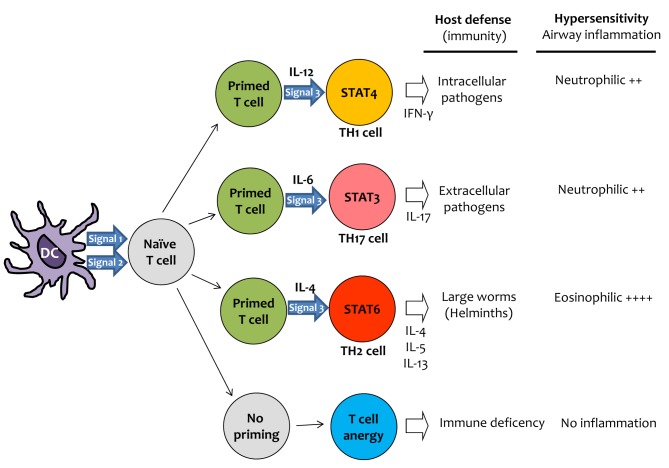 | Fig. 1General scheme of helper T cell priming and polarization. T cell priming requires both signals 1 and 2. Signal 1 is the antigen-specific signal that is mediated by T-cell receptor triggering by MHC class-II-associated peptides processed from antigens. Signal 2 is the costimulatory signal, mediated mainly by triggering of CD28 (by CD80 and CD86 expressed by dendritic cells after ligation of pattern-recognition receptors (PRR), which are specialized to sense pathogens through recognition of pathogen-associated molecular patterns (PAMP). Primed T cells are induced by signal 3 to differentiate into T cells that secrete distinct cytokines. Signal 3 is the polarizing cytokine signal (such as IL-12, IL-6 and IL-4) that promotes the development of Th1, Th17 or Th2 cells, respectively. The nature of signal 3 depends on the activation of a particular PRR by PAMP. Generally, Th1 cells are important for protection against intracellular pathogens, Th17 cells against extracellular pathogens, and Th2 cells against large worms (helminths). Furthermore, T cell responses to innocuous antigens (allergens) are important for the development of chronic inflammation in the airways. The Th2 cell response is related to eosinophilic inflammation, whereas both Th1 and Th17 cells induce non-eosinophilic (or neutrophilic) inflammation.
|
Allergic asthma is associated with a Th2 type of immune response, as Th2 cytokines are known to cause many of the features of the disease.35 Eosinophilia in the airways has long been linked to allergic asthma, and Th2 cytokines, such as IL-4, IL-5, and IL-13, are responsible for eosinophilic inflammation in the airways (Fig. 1).36,37 Although a number of cytokines are involved in promoting B cells to produce antibodies and generate plasma cells, only IL-4 and IL-13 are known to promote isotype switching to IgE.38-40
Th17 cells, a CD4 subset that has emerged within the last 5 years, require both TGF-β and IL-6 for differentiation although these cytokines are not sufficient for differentiation.41 Th17 cells produce mainly IL-17, some IL-22 and IL-21, small amounts of IFN-γ, and no IL-4.42 IL-17 is clearly detectable in inflamed lungs, and IL-17 may be needed for the initial development of airway inflammation in mice; however, administration of IL-17 decreases eosinophilia.33,43,44 Adoptive transfer of Th17 cells induces neutrophilia and imparts resistance to steroid therapy, suggesting that IL-17 may function to promote neutrophil recruitment and could therefore be particularly important in steroid-resistant neutrophilic asthma (Fig. 1).33,45 In addition, IL-17 has been shown to interfere with epithelial cell production of eotaxin,46 which plays a major role in the recruitment and activation of eosinophils.47 Therefore, IL-17 may regulate the eosinophil-neutrophil balance in the lung.
Increased levels of Th1 cytokine IFN-γ were found in the serum, lung tissues, and induced sputum of asthmatic patients. Moreover, the expression of IFN-γ in induced sputum was enhanced in patients with severe asthma compared to those with mild-to-moderate asthma, and correlated with the numbers of sputum neutrophils.32 Transgenic mouse experiments clearly demonstrated that high levels of IFN-γ in airways induce neutrophilic lung inflammation, emphysema48 and AHR.49 In addition, IFN-γ inhibits allergen-induced eosinophil recruitment into mouse lung tissue.18 TGF-β1 is a key mediator in the development of eosinophilic inflammation and tissue remodeling induced by Th2 cells, such as airway fibrosis.50 We showed that TGF-β1 production and eosinophilic inflammation after an allergen challenge are enhanced in IFN-γ-deficient mice.32 Moreover, Wenzel et al.21 found that the airways of patients with severe eosinophilic asthma had greater subbasement membrane thickening and TGF-β1-producing cell numbers than those with non-eosinophilic asthma. Taken together, these findings suggest that IFN-γ is a key mediator in the development of non-eosinophilic severe asthma (Fig. 1).
Go to :
ENDOTOXIN EXPOSURE AS A RISK FACTOR FOR ALLERGIC ASTHMA
The endotoxin lipopolysaccharide (LPS) is a cell wall component of Gram-negative bacteria that is ubiquitous in our living environment. Although human exposure to endotoxin is inevitable, the level of airway exposure is highly variable and presumably correlates with the amount of inhalable endotoxin in house dust particles and air pollutants. LPS concentrations in house dust range from 0.59 to 41.04 ng/mg51; Michel et al.52 indicated that the median LPS concentration in house dust is roughly 2.58 ng/mg; at a mean total density of 171.6 mg/m2 in homes, the mean total LPS density is 446 ng/m2. In a subject inhaling a total of 5 m3, the total LPS exposure at the intrabronchial level is approximately 27 ng over 24 hours in a domestic dwelling.52 When airborne materials like LPS are inhaled, a proportion is rapidly cleared from the nose into the throat by mucociliary action, and this portion of the dose is swallowed. Accordingly, it has been estimated that only 30% of the inhaled dose is actually delivered into the lower airways,53 and the intrabronchial level is approximately 30-ng LPS (defined as the amount to which a subject is exposed at a median LPS concentration during 1 day in an indoor environment).
Epidemiology studies in industrialized countries suggest that a reduced infectious burden (with endotoxin as a marker) is inversely correlated with an increased prevalence of allergic diseases in the population, apparently supporting the 'hygiene hypothesis'.54,55 However, accumulating evidence suggests that endotoxin exposure is inversely associated with developing atopy, but positively associated with an increased risk of asthma and asthma severity in both adults and children.56 Similarly, increasing evidence suggests that the infectious burden in early childhood is positively associated with the development of atopic asthma.57,58 Moreover, patients with neutrophilic asthma had a greater prevalence of airway bacterial colonization and a trend of higher levels of endotoxin in their airways.59 Occupational exposure to dust is associated with an increased prevalence of respiratory disease.60 Several studies revealed that the concentrations of LPS in dust, not dust itself, were positively correlated with decreased pulmonary functions.61,62 Collectively, endotoxin exposure in early childhood may be related to asthma development and/or severity, although this exposure is negatively associated with IgE sensitization to common allergens.
Go to :
MODELING ALLERGIC ASTHMA USING INHALATION OF LPS-CONTAINING ALLERGENS
Airway application of allergen alone was ineffective and inconsistent in eliciting allergic asthmatic responses in mice. The reason was not clear until Eisenbarth et al.63 systematically investigated the effect of contaminating LPS in ovalbumin allergen preparation. In their study, sensitization was carried out by intranasal instillation three times using OVA with no LPS, low-dose LPS (100 ng/mouse), or high-dose LPS (100 µg/mouse). Allergen challenge was performed through intranasal OVA administration alone. LPS-depleted OVA did not induce an inflammatory response in the lung. OVA plus low-dose LPS induced a typical Th2 cell response, including eosinophilic infiltration in the BAL and lung tissue, mucus production, increased serum OVA-specific IgE and IgG1, and draining lymph node (DLN) Th2 cytokine production. On the other hand, high-dose LPS induced a strong Th1 inflammatory response, including neutrophilia, increased IgG2a in the serum, and DLN Th1 cytokine production without mucus. This study provided experimental evidence that the nature of the inflammation depends on the dose of LPS.
We also investigated whether LPS at different doses may lead to different forms of allergen-induced asthma phenotypes, including AHR.32 Our studies demonstrated that OVA with low-dose (100 ng/mouse) LPS induced a Th2 cell response with eosinophilia and allergen-specific IgE production, whereas OVA with high-dose (10 µg/mouse) LPS induced a Th1 cell response with neutrophilia in BAL fluid. More importantly, in both situations after the OVA challenge, AHR to methacholine challenge was significantly enhanced, which is an important component of the allergic asthma phenotype. More recently, we found that OVA with high-dose LPS induced increased expression of Th17 cells and neutrophilic inflammation, which were impaired in IL-17-deficient mice.33 Thus, the neutrophilic inflammation seen in the case of high-dose LPS is actually a mixture of Th1 and Th17 inflammation.
Go to :
IMMUNOLOGIC MECHANISM OF TH2 CELL POLARIZATION BY INHALATION OF LPS-CONTAINING ALLERGENS
The default immune response to inhaled innocuous proteins (allergens) is the development of immune tolerance.64,65 It is well known that IL-4, via STAT6 signaling, is a key mediator of the development of Th2 cell polarization.66,67 We found that airway application of low-dose LPS (100 ng/mouse) enhanced the production of IL-4 during allergen sensitization and induced an allergen-specific Th2 cell response. In addition, the Th2 cell response induced by allergens containing low-dose LPS was impaired in STAT6-deficient mice.32 These findings indicate that IL-4-induced STAT6 signaling during T cell polarization is critical in the development of Th2 cells (Fig. 2).
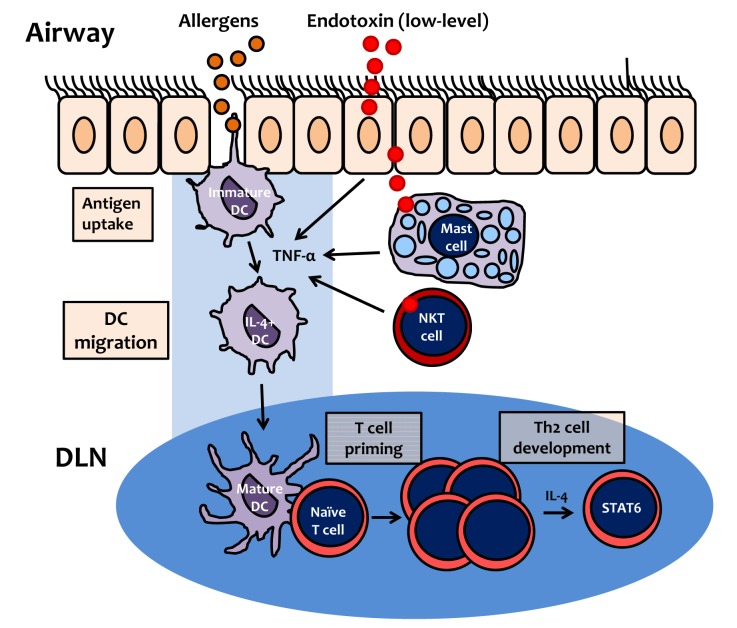 | Fig. 2Proposed mechanism of Th2 cell polarization by inhalation of allergen contaminated with endotoxin (lipopolysaccharide). At low levels, lipopolysaccharide (LPS) induces TNF-α production by airway structural cells, such as epithelial cells, mast cells, or NKT cells. Immature dendritic cells (DCs) uptake allergens, and then migrate to draining lymph nodes (DLN). During allergen uptake by DC, TNF-α, produced in response to low-level LPS, induces DC maturation, expression of costimulatory molecules, and upregulation of DC IL-4 expression. In DLN, mature DCs induce proliferation of naïve T cells (T cell priming), which subsequently differentiate into Th2 cells, via IL-4 and the STAT6 signaling pathway.
|
In terms of the effect of the LPS-induced innate immune response on the development of Th2 cell polarization, Eisenbarth et al.63 showed that TNF-α co-administration with inhaled allergens elicits a Th2 response rather than T cell tolerance. Our data also indicated that allergen sensitization with low-dose LPS induces the Th2 cell response in association with upregulation of TNF-α production during allergen sensitization. Moreover, we found that allergen-specific low-dose LPS-enhanced Th2 inflammation and AHR were impaired in TNFR1-deficient mice.32 Taken together, these findings suggest that TNF-α induced by low-dose LPS is a key mediator of the development of the Th2 cell response to inhaled allergens, mainly via IL-4 and the STAT6 signaling pathway (Fig. 2).
Go to :
IMMUNOLOGIC MECHANISM OF TH1 CELL POLARIZATION BY INHALATION OF LPS-CONTAINING ALLERGENS
It has been well documented that IL-12, produced by the innate arm of the immune system, is an important regulator of Th1 cell development.68-70 We found that LPS dose-dependently enhanced IL-12 expression during allergen sensitization.32 A biological effect of IL-12 is to induce Th1 cell polarization via the STAT4 signaling pathway.71 In the allergic asthma mouse model induced by LPS-containing allergens, we found that the AHR and neutrophilic inflammation induced by sensitization with OVA plus high-dose LPS (10 µg/mouse) were inhibited in STAT4-deficient mice. We also found that lung infiltration of Th1 cells was completely impaired in STAT4-deficient mice.32 These findings indicate that Th1 cell polarization is dependent on the IL-12 induced by exposure to high levels of LPS, which results in IFN-γ production via STAT4 signaling (Fig. 3).
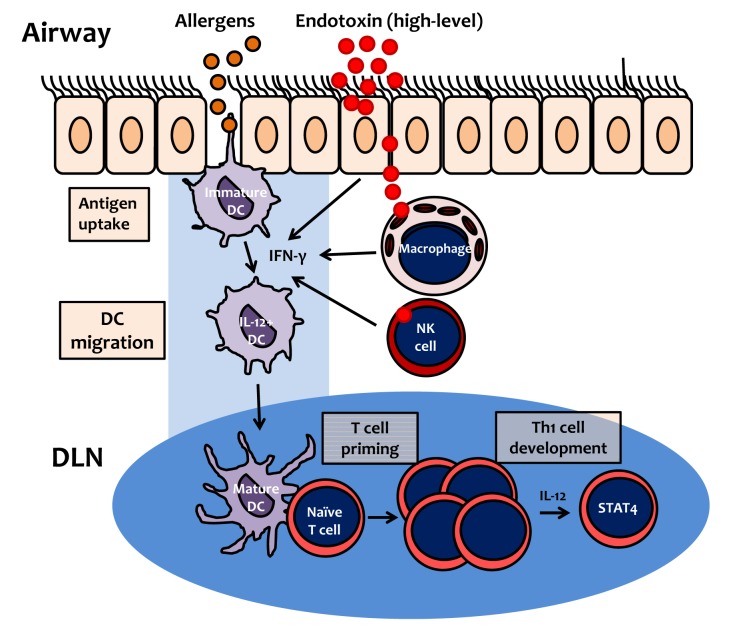 | Fig. 3Proposed mechanism of Th1 cell polarization by inhalation of allergen contaminated with endotoxin (lipopolysaccharide). At high levels, lipopolysaccharide (LPS) induces IFN-γ production by airway structural cells, such as epithelial cells, macrophages or NK cells. Immature dendritic cells (DCs) uptake allergens, and then migrate to draining lymph nodes (DLN). During allergen uptake by DC, IFN-γ, produced in response to high-level LPS, induces DC maturation, expression of costimulatory molecules, and upregulation of DC IL-12 expression. In DLN, mature DCs induce proliferation of naïve T cells (T cell priming), which subsequently differentiate into Th1 cells, via IL-12 and the STAT4 signaling pathway.
|
IFN-γ, a dimerized soluble cytokine, is the only member of the type II class of interferons and is critical for innate and adaptive immunity. IFN-γ is produced predominantly by natural killer (NK) and NKT cells as part of the innate immune response, and by Th1 and cytotoxic T effector cells after development of antigen-specific immunity. IFN-γ promotes Th1 cell polarization by upregulating the transcriptional factor T-bet, which suppresses Th2 cell polarization.18 To summarize, IFN-γ production induced by high-dose LPS upregulates IL-12 expression in antigen-presenting cells, and then the STAT4 signaling pathway mediates a positive feedback loop between IL-12 and IFN-γ during Th1 recall in response to inhaled allergens (Fig. 3).
Go to :
IMMUNOLOGIC MECHANISM OF TH17 CELL POLARIZATION BY INHALATION OF LPS-CONTAINING ALLERGENS
The mechanism by which Th17 cells are polarized from naïve T cells has been studied extensively; IL-6, in synergy with TGF-β, induces IL-17 expression in T cells via the STAT3-RORγt pathway.72 In the allergic asthma mouse model induced by LPS-containing allergens, our data indicate that the production of IL-6 is enhanced by high-dose LPS (10 µg/mouse), but not by low-dose LPS (100 ng/mouse). Moreover, the Th17 cell response induced by an allergen containing high-dose LPS is abolished in IL-6-deficient mice.33 These findings suggest that the enhancement of IL-6 by LPS is a key mediator of the development of Th17 cell polarization, and acts via the STAT3 signaling pathway (Fig. 4).
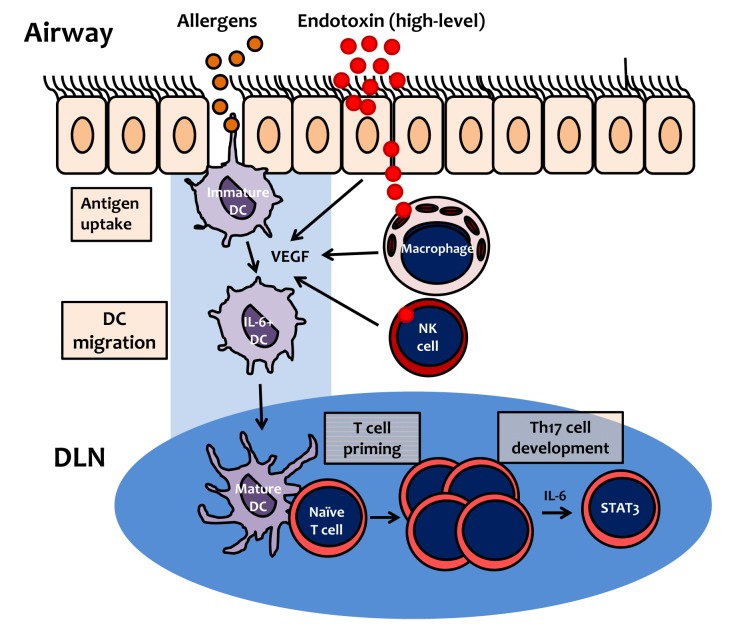 | Fig. 4Proposed mechanism of Th17 cell polarization by inhalation of allergen contaminated with endotoxin (lipopolysaccharide). At high levels, lipopolysaccharide (LPS) induces VEGF production by airway structural cells, such as epithelial cells, macrophages or NK cells. Immature dendritic cells (DCs) uptake allergens, and then migrate to draining lymph nodes (DLN). During allergen uptake by DCs, VEGF (produced by high-level LPS) induces DC maturation and expression of costimulatory molecules via the VEGFR1-dependent pathway. IL-6 expression in DCs is also upregulated by VEGF via the VEGFR2-dependent pathway. In DLN, mature DCs induce proliferation of naïve T cells (T cell priming), which subsequently differentiate into Th17 cells, via IL-6 and the STAT3 signaling pathway.
|
Vascular endothelial growth factor (VEGF) was originally described as a vascular permeability factor based on its ability to generate tissue edema.73 Enhanced levels of VEGF have been detected in tissues and biological samples from patients with asthma.74,75 VEGF is produced during the LPS-induced innate immune response.76 Lung-specific VEGF transgenic experiments revealed that high levels of VEGF in the airways induce lung inflammation and enhance T cell priming to inhalant allergens.77 In the atopic asthma mouse model induced by LPS-containing allergens, we found that airway exposure to high-dose LPS (10 µg/mouse) upregulates the production of VEGF. In addition, recombinant VEGF enhances T cell priming on exposure to allergens alone.33 Moreover, our previous data revealed that T cell priming; e.g., over-expression of costimulatory molecules on dendritic cells (DC) and DC migration into regional DLN, and subsequent proliferation of naïve T cells in DLN, are blocked by treatment with a VEGFR1 inhibitor in the atopic asthma model. However, pharmacological intervention with a VEGFR-2 inhibitor abolished the LPS-induced production of IL-6 (but not IL-12p70), and the subsequent development of an allergen-specific Th17 cell response.78 Together, these findings suggest that LPS-induced VEGF is a key mediator of the development of T cell priming and the Th17 cell response to inhaled LPS-contaminated allergens. In addition, T cell priming to LPS-containing allergens is dependent on VEGFR1-mediated signaling, and the subsequent Th17 polarization is dependent on on VEGFR2 signaling (Fig. 4).
Go to :
CONCLUSIONS
Biological contaminants in indoor air can induce immune dysfunction and inflammation, resulting in inflammatory pulmonary disorders, such as asthma and chronic obstructive pulmonary disease (COPD).32 A recent nationwide survey indicates that household LPS exposure is a significant risk factor for increased asthma prevalence.79 Asthma prevalence has been increasing over recent decades in developed countries, particularly in urban areas.80 This increase in asthma prevalence may be related to increased indoor activity. Homes insulated from outside exposure, such as apartments in urban areas, contain greater concentrations of biological contaminants. This change in living environment may be related to the increased asthma prevalence due to enhanced exposure of the airway to the biological contaminants such as house dust mite-derived allergens and bacterial products (including LPS) present in indoor dust.
New therapeutic approaches may stem from a greater understanding of immunopathogenesis of asthma. Mild and moderate asthma may be related to eosinophilic inflammation, whereas severe asthma is associated with neutrophilic inflammation.11,82 In terms of immunopathogenesis, eosinophilic asthma represents Th2 cytokine-dependent inflammation, whereas neutrophilic asthma is related to both IFN-γ- and IL-17-dependent inflammation.33,82,83 In view of the different immune and inflammatory patterns of atopic asthma, it is not surprising that responses to anti-inflammatory therapies differ: mild-to-moderate eosinophilic asthma is usually highly responsive to inhaled corticosteroid therapy, whereas patients with neutrophilic (severe) asthma respond poorly. Future therapeutic strategies for neutrophilic asthmatic patients may be aimed at inhibiting corticosteroid resistance. To achieve this aim, suppression of Th1 and Th17 cell responses and inflammation may be a useful approach to asthma therapy.
Go to :
 XML Download
XML Download