

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The term "atopy" refers to a family of diseases including eczema, asthma and allergic rhinitis. Atopic dermatitis (AD) is a chronic inflammatory skin disease evolving from a genetic defect in proteins relevant for the epidermal barrier in some patients. Still, all patients suffering from AD show an impaired cutaneous barrier function. Most often, patients present in early childhood with dry skin and relapsing eczema. During the following years additional food allergies, allergic rhinitis and asthma may appear, a process known as the "atopic march". In industrialized countries atopic diseases are very common, 15%-30% of all children and 2%-10% of adults are affected and epidemiological studies suggest that atopic diseases increased significantly in most westernized countries during the last decades. Currently two major models exist to explain the pathogenesis of AD: The predominant model characterizes AD as a result of an impaired epidermal barrier function due to intrinsic structural and functional abnormalities in the skin. In this model the impaired epidermal barrier is the primary problem.1 The second and traditional model views AD primarily as an immune function disorder in which Langerhans cells, T cells and other immune effector cells modulate an inflammatory response. Widely accepted for years, there is now lesser support for this theory and probably an impaired barrier and a compromised adaptive immune answer both contribute to the disease.2,3
Intact surface barriers are absolutely necessary and vital for the survival of living organisms. Whereas unicellular organisms are enclosed by cell membranes and cell walls, the surface of multicellular organisms is covered by epithelial barrier structures. The epidermis of mammalians consists of a single layer of proliferating cells, the stratum basale, and several superficial layers of stratum spinosum und stratum granulosum (SG), which finally form the stratum corneum (SC). An intact epidermis prevents environmental irritants, allergens, and microbes from entering the body. Permeability of the epidermis is determined by complex interactions of differentiated keratinocytes on the surface of the skin and structural proteins, such as filaggrin, regulatory enzymes and lipids. Any disruption of these components through inherited defects, trauma, decreased humidity, alteration of pH, and infection can decrease the ability of the epidermis to function as an effective barrier. Disruption allows antigenic and irritant agents as well as allergens to penetrate the barrier and come into contact with immune cells, leading to a release of pro-inflammatory mediators. This can induce the above described typical clinical and pathological findings of AD or other immune disorders such as food allergies, allergic rhinitis, asthma or inflammatory bowel disease.2,4
THE EPIDERMAL BARRIER IN ATOPIC DERMATITIS
The healthy epidermis consists basically of three different barriers: the tight junctions as a liquid-liquid interface barrier, the squamous cells as an air-liquid interface barrier and immunocompetent cells such as the Langerhans cells as an immune barrier.5 In vertebrates tight junctions (TJ) act as a barrier to demarcate different fluid compartments. TJs exist at body surfaces, such the intestine, trachea, and the stratified epithelia of the skin. The three stratum granulosum layers are called SG1, SG2, and SG3,6,7 counting from the surface inward. When SG3 cells differentiate into SG2 cells, they form TJs and begin to secrete lamellar granules from their apical membranes into the extra-TJ compartment. These granules contain lipids, extracellular structural proteins, hydrolytic enzymes and various antimicrobial peptides (AMPs) such as cathelicidin and β-defensins.8 After SG2 cells differentiate into SG1 cells they loose their TJs and undergo final cornification. Langerhans cells (LC) are epidermal dendritic cells that are specialized in antigen presentation. LCs are located close to the TJ barrier aiming their dendrites outward. LCs are activated by TSLP, TNF-α, and IL-β, presumably secreted from damaged keratinocytes and subsequently extend their dendrites through TJ barriers and take up antigens from the extra-TJ environment to induce a T helper cells 2 (Th2) immune responses. Taken together the SC, TJs and LCs together constitute a very effective skin surface surveillance and barrier system.
Stratum corneum dysfunction contributes to percutaneous sensitization
The epidermal barrier can be altered by defects in the structural barrier, the peptide barrier or the immunological barrier. Mutations in the filaggrin gene (FLG) are predisposing factors for atopic diseases. Filaggrin monomers display keratin-binding activity in vitro and have been proposed to induce the compaction of corneocytes by contributing to keratin pattern formation in the lower SC. Filaggrin monomers become degraded into natural moisturizing factor (NMF) to maintain hydration of the upper SC and to reduce the pH of the skin surface.9 FLG mutations were identified initially as a cause of ichtyosis vulgaris and as a major predisposing factor for AD. Furthermore, they have been reported to be associated with atopic asthma, allergic rhinitis, nickel and food allergies,10,11 suggesting that FLG mutation associated SC barrier defects lead to increased numbers of episodes of percutaneous allergen exposure. Interestingly FLG mutations are not associated with asthma without eczema,10,11 and most of the identified asthma-associated genes are not associated with AD suggesting atopic asthma as a sub-entity of asthma. Because filaggrin is not expressed in the upper airways systemic sensitization is likely to occur due to percutaneous antigen exposure through filaggrin-deficient skin.12
To induce epicutaneous sensitization to antigens the SC barrier has to be mechanically impaired by tape stripping, acetone treatment, or patch dressing. Thus, perturbation of the SC barrier not only allows allergen penetration throughout this barrier but also triggers LC activation and facilitates subsequent uptake of antigens by LCs across the epidermal TJ barrier. After antigen acquisition, LC migrate to draining lymph nodes and activate antigen-specific T cells.4,13 Further allergens and microbial factors that have penetrated defective skin barriers induce inflammation while inflammation itself can alter skin barrier integrity. Th2 and Th17 cytokines have been reported to downregulate filaggrin expression or can alter processing of profilaggrin.14-16 Due to the fact that not only genetic skin barrier defects, but also genetic immune disorders such as Wiskott-Aldrich syndrome present atopic manifestations it seems reasonable that AD results from an interplay between both, epidermal barrier and immunity-associated genetic dysfunction. Taken together it seems reasonable to hypothesize that immunity-associated genetic factors as well as environmental or microbial factors may act additively to produce SC barrier defects and to promote percutaneous sensitization during the onset of AD.5
Antimicrobial peptides in the skin's innate immune defense
The human skin forms the initial defense barrier against invading microbial pathogens. Professional innate immune cells such as DCs and macrophages battle infections. Also small cationic peptides, coined AMPs contribute to the chemical shield on the surface of the skin and other epithelia. AMPs are a diverse group of structurally distinct peptides with similar functions. So far several dozens of different peptides with antimicrobial function in the skin are known.17 Unfortunately, no clear definition for antimicrobial activity exists so that the list of skin derived AMPs is expected to grow. It is known that AMPs are not only endogenous antibiotics that can destroy bacteria, viruses and fungi but also that AMPs can act as immune modulators with impact on innate and adaptive immune functions.18 In addition to keratinocytes also cells in the eccrine glands, mast cells and sebocytes produce and secrete AMPs. Furthermore invading immune cells, such as neutrophils and NK cells contribute to the pool of AMPs in the skin.8,18-20
The probably best studied AMP gene families in skin are the defensins and cathelicidin.21,22 The first skin derived AMP found in humans was β-defensin 2 (HBD2).23 HBD2 is activated by skin inflammation, skin infection and ultraviolet light B (UVB) irradiation and is very effective against gram-negative bacteria.24 A second well-studied cutaneous AMP is cathelicidin, often referred to its peptide form hCAP18 or LL-37. Similar to HBD2, cathelicidin is inducible by UVB irradiation in vivo.25 The human genome contains only a single cathelicidin gene (CAMP). Cathelicidin is produced as a pro-peptide which is composed out of an N-terminal cathelin domain and a C-terminal amino sequence with antimicrobial activity. This part must be sliced off from the inactive precursor by serine proteases of the kallikrein family to form active LL-37.22,26 Later cathelicidin can be processed to smaller peptides with different functions.27 In solution, LL-37 forms an α-helix enabling these peptides to destroy bacterial membranes, viral envelopes and candida.17,28 Furthermore LL-37 can interact with mammalian cells in a ligand-receptor mediated or a receptor-independent manner resulting in a host response which is called the "alarmin" function of LL-37. Alarmin functions include the direct interaction with the formyl-peptide receptor like 1 or G-coupled receptors.26,29 Furthermore, cathelicidin influences adenosine triphosphate (ATP) receptor P2X7 and toll-like receptor (TLR) signaling in immune cells, epidermal growth factor (EGF) receptor transactivation and intracellular Ca2+ mobilization. Also, chemokine and cytokine release can be induced by cathelicidin.30 In synergy with the immune modulator IL-1β, LL-37 enhances innate immune responses by multiple pathways.18,31 Taken together, the antimicrobial and alarmin function of cathelicidin underscores the importance of this peptide in cutaneous innate immune defense. Disturbed function of cathelicidin on the other hand could contribute to the pathogenesis of skin disease.
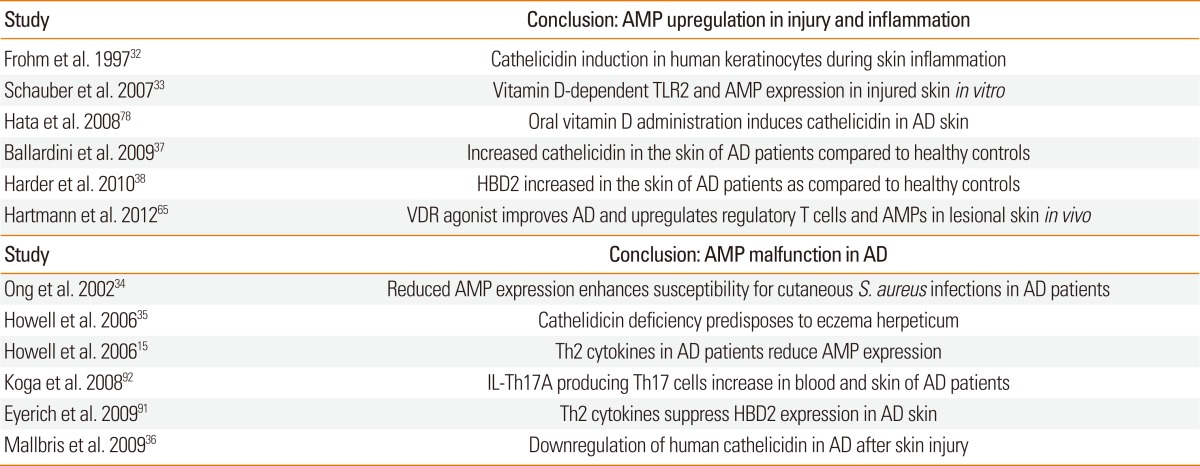
In healthy skin cathelicidin and other AMPs protect the skin from microbial infection. Physiologically, only very low amounts of cathelicidin can be detected. Multiple studies reported an increase of cathelicidin in cutaneous infections and wounds.32,33 In some skin diseases where barrier functions of the skin and control of inflammation is disturbed cathelicidin levels are altered compared to healthy skin. Already a few years ago, it has been hypothesized that the reason for the common viral or bacterial infections in AD patients might be due to disturbed AMP expression.34 An altered cytokine milieu was suggested to reduce cathelicidin expression and other AMPs such as human β-defensin 2 (HBD2). In particular, Th2 cytokines such as interleukin-4 (IL-4) and interleukin-13 (IL-13) suppress the induction of AMPs.15 Also, in a subgroup of patients suffering from infectious complications the expression of cathelicidin AMP is even lower than in AD patients without skin infections.35 Consequently, the pathways which increase cathelicidin in healthy individuals might be impaired in AD patients.36 However, other studies found increased cathelicidin protein in lesional skin of patients with AD compared to non-lesional skin or skin from healthy controls.37 Also, an increased expression of HBD2 and other AMPs in lesional skin of patients with AD as compared to healthy controls was reported.38 Thus, up to present it is not completely clear whether cathelicidin or other AMPs contribute to the pathogenesis of AD. However, as the mechanisms regulating AMP expression in the skin become clearer these peptides could still be targets for supportive, barrier-enhancing therapies. Unexpectedly, one of the pathways involved in AMP regulation in skin could be the vitamin D pathway.
VITAMIN D AND IMMUNE FUNCTIONS
Vitamin D deficiency (as determined by 25(OH)D serum levels) is very common and only two-thirds of the American population show sufficient vitamin D serum levels of 50 to 125 nmol/L. About one-quarter of the American population is at risk of vitamin D inadequacy (30-49 nmol/L), and 8% were at risk of vitamin D deficiency (<30 nmol/L). Other studies report that up to 30% of the adult Western population and up to 70% of the elderly institutionalized patients suffer from hypovitaminosis D.39 Sparse sun exposure and an unbalanced and insufficient diet are thought to be reasons for low vitamin D serum levels. Hypovitaminosis D is assumed to contribute to increased susceptibility to infections and a higher mortality risk.40 Since the beginning of the 20th century it is known that sufficient intake of vitamin D is important for calcium homeostasis and bone health. Age, female sex, higher latitude, winter season, darker skin pigmentation, less sunlight exposure, dietary habits, and absence of dietary vitamin D fortification are the main factors that are significantly associated with low vitamin D serum levels.39
Nevertheless, under normal circumstances the human body is able to synthesize sufficient vitamin D from precursors under the influence of UVB irradiation.41 1,25-dihydroxyvitamin D, the hormonally active form of vitamin D, is produced by the interplay of different cell types and organs. Under the influence of UVB irradiation in the range of 280-320 nm the precursor 7-dehydrocholesterol (provitamin D) is chemically modified in the skin.42 The result of this fast photochemical process is calciol (previtamin D), an inactive form of vitamin D. Previtamin D is then transported to the liver by vitamin D binding protein. In the liver calciol gets hydroxylated by the vitamin D 25-hydroxylase (CYP27A1). Hydroxylation of 25-hydroxyvitamin D 1-α-hydroxylase (CYP27B1) happens then in the kidney where active 1,25-dihydroxyvitamin D (calcitriol) is synthesized. Independent of this mechanism, keratinocytes of the basal and suprabasal layers of the skin express CYP27A1 and CYP27B1 enabling a direct synthesis of 1,25-dihydroxyvitamin D from the precursor 7-dehydrocholesterol (provitamin D).43 As vitamin D is required for normal keratinocyte proliferation, differentiation and function, this mechanism is of special importance.42-44 As a consequence, a disturbed or insufficient vitamin D metabolism could directly influence keratinocytes and their intrinsic functions.44
The influence of vitamin D on the cutaneous innate immune system
During the last years vitamin D has become the subject of intensive research as an immune regulator both of the innate and adaptive immune system.41,45 A powerful defense against microbial or other pathogens at the cutaneous surface has to be quick in action and fine tuned in process to avoid excessive inflammation and tissue damage. Cells of the innate immune system recognize danger signals via toll-like receptors. Pathogen-associated molecular patterns (PAMP), which exist mainly as lipopolysaccharides (LPS) on microbial cell membranes, or in microbial or viral proteins and deoxyribonucleic acid (DNA) are sensed by TLRs. Activation of innate immunity cells then induces secretion of inflammatory mediators finally recruiting additional cells of the innate and adaptive immune system. Interestingly, vitamin D affects different aspects of innate immunity. On the one hand, vitamin D inhibits a pathologic inflammatory response by suppressing the TLR-mediated inflammation on DCs. Here, vitamin D decreases the expression of immune receptors and co-stimulatory molecules inhibiting dendritic cell activation by LPS. Furthermore cytokine secretion declines and leads to reduced antigen presentation. Also, DC differentiation, maturation, chemotaxis, and antigen presentation seem to be dampened. On the other hand, vitamin D helps to defend opportunistic infections by inducing autophagy in human macrophages and to support the innate skin barrier by stimulating endogenous AMP expression in resident epithelial cells of the skin and lung.33,46,47
The influence of vitamin D on the adaptive immune system
The adaptive or specific immune system is characterized by its ability to recognize known and adapt to new pathogens. The adaptive immune response involves lymphocytes with antigen-specific receptors recognizing and eliminating foreign pathogens. T-cells contain the surface marker cluster of differentiation (CD) 3 and can be divided into CD8+ cytotoxic T-cells (CTL) and CD4+ T-helper cells (Th). Th cells are further characterized by specific cytokine secretion patterns and divided in Th1, Th2, Th9, Th17 and Th22 cells. Almost all cells of the adaptive immune system express the vitamin D receptor (VDR) which renders these cells easily responsive to vitamin D.45 As a consequence, vitamin D decreases pro-inflammatory cytokine released from peripheral blood mononuclear cells (PBMC) particularly from T cells.48 In addition, vitamin D inhibits T cell proliferation through decreased Th1 cytokine secretion.49 The effects on Th2 cells are less clear. Whereas one study describes vitamin D-induced IL-4, IL-5 and IL-13 in vitro other studies does not show any effect. Regardless, vitamin D supplementation does not induce Th2 responses in vivo. Furthermore, pro-inflammatory Th17 responses are also blocked by administration of vitamin D in mice and man. Also, vitamin D increases IL-10 levels and decreases the production of IL-2 thereby inducing a state of hypo-responsiveness in regulatory T cells (Treg) cells. Such hypo-responsiveness is usually seen with corticosteroid treatment.48 Recently, it has been shown that vitamin D application leads to an inhibition of effector T cells and vitamin D deficiency may promote autoimmunity by favouring the excessive production of Th17 and Th9 cells at the expense of IL-10-producing Treg. Furthermore, vitamin D suppresses adaptive immunity by inhibition of antigen presenting cells.50 Upon vitamin D treatment of T-cells, Th2 cytokines such as IL-4, IL-5 and IL-10 were increased whereas IL-2 and IFN-γ, typical Th1 cytokines needed for T-cell proliferation and activation, were decreased.49,50 Additionally a murine model showed that Th17 cell functions were inhibited while regulatory T cells (Treg) were increased upon vitamin D treatment.51 Finally, vitamin D affects T-cell homing in the skin via expression of the chemokine receptor 10 (CCR10). CCR10 and its skin-associated chemokine CCL27 mediate chemotactic responses of cutaneous memory T cells that express the CCR10. In this context, the expression of CCR10 on T-cells was increased upon vitamin D treatment. As CCL27 is selectively expressed by keratinocytes in the epidermis vitamin D is thought to mediate the chemoattraction of T-cells and the "epidermotropism" of CCR10+ T cells from the underlying dermal layer through increased CCR10.52
In addition to T lymphocytes, B cells seem also be targets of vitamin D: B lymphocytes can recognize foreign antigens via their B-cell receptor (BCR). Subsequently activated B-cells proliferate to antigen-producing plasma cells or memory cells. These plasma cells produce soluble antibodies. Remarkably, B-cell proliferation and immunoglobulin production can be inhibited by vitamin D. Vitamin D also effects B lymphocyte functions and modulates the humoral immune response including secretion of immunoglobulin E (IgE).
Other allergy mediating cells, e.g. mast cells and eosinophils, are also targets of vitamin D. Enhanced cutaneous vitamin D synthesis increases the production of IL-10 in mast cells leading to suppressed skin inflammation. Additionally, vitamin D-treated mice showed reduced airway hyperresponsiveness and decreased infiltration of eosinophils in the lung.53 However, only pre-activated, proliferating B-cells were affected whereas the initial B-cell division was not affected.
In sum, vitamin D has pleiotropic effects on immune functions in the skin which are relevant for the development of atopic diseases such as AD and allergies.
THE ROLE OF VITAMIN D IN ALLERGIC DISEASES
As outlined above, vitamin D plays an important role in the regulation of our immune functions. In particular, vitamin D regulates the activity of various immune cells, including monocytes, dendritic cells (DCs), T and B-lymphocytes, as well as immunological functions of epithelial cells. Furthermore, some immune cells express vitamin D-activating enzymes facilitating local conversion of inactive vitamin D into active calcitriol with subsequent paracrine and autocrine effects. Unfortunately both, atopy and hypovitaminosis D, show a high prevalence in the general population making statistical examinations very difficult.39 As described above vitamin D significantly affects inflammatory pathways and cells involved in the development and course of allergic diseases. Consequently, several studies have investigated a relationship between vitamin D and allergic diseases.
The clinical impact of vitamin D on allergies in adults
Wjst and Dold54 hypothesized already in 1999 a link between nutritional intake of vitamin D and allergies. In 2004, Zitterman et al. then suggested that vitamin D deficiency may lead to an increased risk of allergies when observing an association between low vitamin D status and low levels of the tolerogenic cytokine IL-10 cord blood. Later, Camargo and coworkers55 suggested that regional differences in vitamin D status were responsible for a strong north-south gradient for the prescription of adrenalin auto-injectors as emergency treatment for severe allergic reactions in the United States. In adult patients several clinical studies then analysed vitamin D levels and the severity of allergies such as asthma and chronic rhinoconjunctivitis (CRS). While in one study no difference in serum vitamin D levels of adult asthma patients and healthy controls was observed, Li and coworkers56 found that vitamin D deficiency was highly prevalent in allergy patients and vitamin D status strongly correlated with pulmonary function (FEV1) in asthmatics. Also other observations showed that higher vitamin D serum levels improve pulmonary function in adults while insufficient serum levels are associated with severe pulmonary dysfunction.57 As vitamin D enhances the effects of glucocorticoids on peripheral blood mononuclear cells from asthmatic patients, corrected
serum vitamin D levels in asthmatic patients could lead to improved disease outcome.58 Interestingly, Hyppönen and coworkers59 showed in patients with low vitamin D (<25 nmol/L) and with very high vitamin D serum levels (>135 nmol/L) significantly higher IgE levels than in healthy individuals (100-125 nmol/L). Correction of serum concentrations of vitamin D reduced IgE level significantly.59,60 For other allergic diseases such as CRS current clinical studies have shown that CRS patients had vitamin D serum levels that were about 40%-50% lower than the vitamin D serum levels in the control group.57,61 In contrast, the incidence of allergic rhinitis was shown to correlate with increased vitamin D serum levels in very early studies from 1930 and 1962.62 Notably, corrections for geographical region and month of examination did not change this association in those studies. A current review of the literature did not show a positive correlation between vitamin D intake and an increased risk of adverse events. However, most studies using higher doses of vitamin D were not adequately designed to assess long-term harms. During the last years multiple studies showed the impact of vitamin D on AD. One of the first randomised controlled trials (RCT) showed a significant SCORing Atopic Dermatitis (SCORAD) reduction after an oral supplementation consisting of vitamin D and vitamin E. However, an exclusive vitamin D treatment did not show any benefit over placebo administration.63 Recently, a RCT including sixty AD patients reported a significant improvement in disease severity in patients with mild, moderate and severe AD taking oral vitamin D as compared to placebo-taking patients.64 Studies of Hartmann et al.65 suggested that endogenous AMPs in the skin of AD patients might be upregulated by oral vitamin D supplementation and thereby ameliorate allergen-triggered eczema. In sum, several studies suggest that vitamin D has beneficial effects on the course of allergic disease. However, the exact mechanisms are so far not well defined.
The clinical impact of vitamin D on allergies in children
The question if the predisposition to allergies might be acquired in utero or during the development of the immune system has been discussed for a long time. Several studies have investigated how and if the intake of vitamin D during maternity or infancy influences the allergy risk in children. However, unfortunately these studies do not draw a clear picture.
First, multiple studies suggested a positive correlation between the intake of vitamin D during maternity or infancy and the prevalence of allergies in children. Camargo et al.66 reported that high vitamin D levels during maternity decreased childhood wheezing in about 50% compared to low maternal vitamin D levels. They also suggested that cord-blood levels of vitamin D are inversely associated with the risk of respiratory infections and childhood wheezing but not with incident asthma. This hypothesis could be reproduced in a larger study which demonstrated that maternal vitamin D intake during pregnancy decreased the risk of wheeze symptoms in early childhood.67 A large metaanalysis of 11 databases confirmed that high maternal dietary vitamin D and E intake during pregnancy seems to be protective against the development of childhood wheezing.68 Another study reproduced those results and reduced maternal intakes of vitamin E, vitamin D and zinc during pregnancy were associated with increased wheezing outcomes in children.69 It seems probable that maternal vitamin D intake during pregnancy increases the mRNA levels of the leukocyte receptors ILT3 and ILT4. Measured in umbilical cord blood increased levels of ILT3 and ILT4 are critical for the generation of T suppressor cells and induction of immunological tolerance. Taken together this finding may point towards an early induction of tolerogenic immune responses by maternal vitamin D intake in the developing child.70 Furthermore, several studies analysed vitamin D serum levels in children suffering from allergic diseases. Still, in children and adolescents allergic sensitization to 11 of 17 investigated allergens was more common in those with vitamin D deficiency. In particular, vitamin D levels below 15 ng/mL were associated with peanut, ragweed and oak allergy. In children with AD mean vitamin D levels were significantly higher in patients with mild disease compared to those with moderate or severe AD.71 Moreover, Mullins et al.72 showed significantly higher rates of food allergy in children born autumn/winter (compared to spring/summer) suggesting a relationship between relative food allergy rates and monthly UV irradiation. They hypothesized that UV light exposure and thereby vitamin D status may be one of many potential factors contributing food allergies in childhood. Finally, a multiple hit model was suggested in which the lack of vitamin D impairs the epithelial barrier integrity leading to increased and inappropriate mucosal exposure to food antigens. In this model, vitamin D deficiency would also promote a pro-sensitization immune imbalance that compromises immunologic tolerance. The authors hypothesized that early correction of vitamin D deficiency might promote mucosal defense, maintain a healthy microbial barrier and allergen tolerance and thereby limit food allergies in children.73
Concerning childhood asthma, it has been published that children with asthma had low serum vitamin D levels independently of the geographical latitude. Of 616 children with asthma 21 (3.4%) had a vitamin deficiency (<20 ng/mL) and additional 152 (24.6%) showed a vitamin D deficiency (20-30 ng/mL).74 Recently a prospective study showed that vitamin D supplementation in children with asthma reduces the risk for recurrent respiratory infections and disease exacerbation.75 Furthermore, vitamin D supplementation in children during the winter months reduced the rate of influenza A infections and the frequency of asthma attacks.76 Also, a recent pilot RCT demonstrated a positive effect of vitamin D supplementation on AD symptoms in children during winter months.77 This effect might be mediated by the induction of endogenous AMPs in the skin in AD by oral vitamin D supplementation.78
Even though these multiple studies favored a strong relationship between vitamin D deficiency and allergies, high vitamin D intake might be harmful. Interestingly, children whose mothers had a vitamin D concentration during pregnancy greater than 75 nmol/L showed an increased risk of AD on examination at 9 months and asthma at the age of 9 years compared to children whose mothers had a concentration of <30 nmol/L.79 Additionally, Hughes and coworkers80 found no association between UV exposure, vitamin D level and childhood asthma. Rather it was shown that UV exposure in the wintertime between the ages 6-15 year increased the probability of having hayfever. Noteworthy, children with vitamin D supplementation (>13.1 µg/day) showed an increased risk to develop AD, allergic rhinitis or allergic asthma.81 In particular, vitamin D intake led to an increased risk to develop AD at the age of 6 if a positive family history for AD was already reported. Similarly, dietary intake of vitamin D during infancy promoted allergic disease at age 31.82 Also another study found that early vitamin supplementation in children was associated with increased risk for asthma and food allergies.83 All of these results have prompted the question whether the mode of application of vitamin D might be also important for promoting allergies. Indeed, Kull et al.84 could show that vitamin D offered in water soluble form seemed to increase the risk of allergic disease up to the age of 4, compared with patients that received vitamin D in peanut oil.
Taken together there exist conflicting data about the interaction of vitamin D, AD, food allergies, allergic rhinoconjunctivitis, and asthma. As far as we know, both very high and very low levels of vitamin D seem to be risk factors for atopic disease. A large metaanalysis of the Cochrane Collaboration was unable to find convincing evidence of dietary supplements in eczema. They considered all of the studies as too small in size and of too low in quality to exclude even moderate treatment benefits. Interestingly, many of the as "placebos" used substances, such as olive oil might not be inert, thereby reducing the chances of finding a true biological effect.85
IS THERE A LINK BETWEEN VITAMIN D, ANTIMICROBIAL PEPTIDE EXPRESSION AND ATOPIC DERMATITIS?
Like other AMPs, human cathelicidin is expressed by keratinocytes, other innate immune cells and cells of the adaptive immune system as described above.19,86 Cathelicidin expression is upregulated in infected and injured skin32 and can induce neovascularization, wound healing, and chemokine expression and migration of immune cells, such as neutrophils, mast cells, monocytes and T-cells.86 The regulation of cathelicidin gene expression was unclear for a long time since infection mediators did not influence gene expression. Several reports eventually confirmed a connection between vitamin D and AMP expression in keratinocytes. Wang et al.46 demonstrated that vitamin D plays an important role in antimicrobial cutaneous immunity when identifying a vitamin D response element (VDRE) in the promoter region of the cathelicidin gene. Soon thereafter, other groups confirmed that cathelicidin constitutes a direct target of vitamin D in keratinocytes.51 Furthermore, it has been shown that the cathelicidin gene is regulated by the vitamin D pathway in humans and non-human primates.87 So far multiple elements in the molecular mechanisms of vitamin D mediated cathelicidin expression have been characterized: vitamin D-induced epigenetic changes, such as histone acetylation and co-activator activity enable the expression of cathelicidin.88,89 Thus, a direct connection between vitamin D metabolism and cutaneous innate defense function was established.
Interestingly, therapies targeting vitamin D such as UVB irradiation can ameliorate skin inflammation in AD. So far, effects of this therapy were attributed to the effect of UVB on T-cells. Nevertheless, the beneficial effects were could also be the result of UVB induced activation of AMPs such as HBD2 and 3 or the UVB induced vitamin D synthesis and subsequent cathelicidin expression in skin.90 IL-17A producing Th17 cells were reported to be increased in the skin and blood of patients with AD91,92 and IL-17A enhances vitamin D induced expression of cathelicidin in keratinocytes.93 Consequently, it was suggested that topical treatment or oral supplementation of vitamin D might improve barrier function and thus inflammation in AD. In one of the few studies investigating this hypothesis increased cathelicidin expression in skin biopsies in patients suffering from AD following a course of oral vitamin D supplementation was observed.78 In a recent study a Japanese working group could furthermore show that patients with AD have low vitamin D serum levels which correlate with low serum levels of cathelicidin peptide.94 Another group recently demonstrated that systemic low-dose treatment with a low-calcemic VDR agonist might improve allergen-induced asthma in vivo. Treatment with this VDR agonist upregulates forkhead box P3 (Foxp3) expressing regulatory T cells in lesional skin and resultes in a robust induction of several skin barrier genes and, moreover, the AMPs β-defensin 2 and β-defensin 3.65 Thus, increasing vitamin D synthesis and elevating vitamin D levels in the serum may help to strengthen the innate cutaneous defense barriers to prevent cutaneous infections which could serve as cofactors for allergic sensitization (Table).
CONCLUSIONS
While there is a growing body of evidence that vitamin D plays a significant role in both, cutaneous AMPs expression and the pathogenesis of allergic diseases the extent of the interplay of both has not been fully elucidated. Clinical studies assessing a possible link between vitamin D and allergies suggest that vitamin D could be beneficial during pregnancy and might prevent allergic skin diseases in children. However, while experimental and pre-clinical data point towards a protective role of vitamin D the clinical results available to date draw a less clear picture. Larger, prospective trials in patients with AD testing the effect of vitamin D supplementation are missing. On a molecular level increased vitamin D could lead to increased barrier function by enhancing antimicrobial defense mechanisms in individuals with AD. After a vitamin D response element (VDRE) was discovered in the cathelicidin gene it was shown conclusively that vitamin D enhances the expression of the AMP cathelicidin in keratinocytes with epigenetic changes and activation of the Act1 and MEK-ERK pathway as co-regulators. Vitamin D could thus enhance barrier function in the skin and ameliorate atopic skin disease. Unfortunately, no large-scale prospective clinical studies determining the effects of vitamin D on cathelicidin expression and on allergic diseases in parallel have been conducted yet. Most of the few studies examining the influence of vitamin D expression on allergies studied AD patients and showed conflicting results. Further randomized controlled trials regarding treatment with vitamin D in context of cathelicidin expression in the skin of patients with allergic skin diseases may help determining a definitive link.
 XML Download
XML Download