

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Allergic rhinitis (AR) and asthma are both allergic diseases of the upper and lower airways that appear to have similar pathophysiologic features.1,2 Common characteristics of these diseases include variable degrees of airflow obstruction, airway hyper-responsiveness, and inflammation in response to allergens. In addition, chronic inflammatory reactions caused by allergens induce significant changes in the structural components of the airway wall, collectively known as remodeling.
Airway remodeling is a central feature of asthma, and it has been demonstrated by a number of studies.3-5 However, there has been much debate about whether such remodeling occurs in AR. Recent studies have revealed that airway remodeling can occur in the nasal mucosa, even though the pathologic extent of nasal remodeling might differ from that of the bronchus.6,7
Airway remodeling results in structural changes that include smooth muscle hypertrophy, goblet cell hyperplasia, subepithelial fibrosis, inflammatory cell infiltration, and increased vascularity.8 Despite the fact that increased vascularity - also called vascular remodeling - is a crucial step in the pathogenesis of the airway remodeling process in asthma,9 it has been largely overlooked in studies of AR. Histological examinations have revealed a significant increase in the number of microvessels in the airways of both pediatric and adult asthma patients.10,11 Vascular remodeling has also been demonstrated in a rat model in response to chronic allergen exposure.12 In addition, nasal vasodilatation and increased vascular permeability are important features of AR, although the underlying mechanisms are unknown.
Vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) are potent angiogenic factors that also act as proinflammatory cytokines by increasing endothelial cell permeability and promoting the upregulation of endothelial cell adhesion molecules.13 VEGF plays a key regulatory role in the formation of blood vessels by controlling the proliferation and migration of endothelial cells. The action of VEGF depends on its interaction with PDGF-B during the stabilizing process of vascular walls.14 Increased expression of VEGF and PDGF is a well-documented feature of asthma.11,15 Of note, VEGF is also known to mediate vascular and extravascular remodeling and inflammation in the lung.16 In addition, administration of anti-PDGF neutralizing antibody significantly reduces airway wall thickening induced by allergen challenge.17 Matrix metalloproteinases (MMPs) also play important roles in airway remodeling caused by VEGF.18 A close relationship exists between VEGF and MMP-9 expression in the sputum of asthma patients, and inhibition of VEGF receptors downregulates the expression of MMP-9 in murine models of asthma.
Therefore, it is hypothesized that angiogenic factors, such as VEGF and PDGF, and associated MMPs are responsible for nasal airway remodeling in AR. However, there have been few studies to elucidate their precise functions. This study was conducted to investigate the role of VEGF and PDGF in nasal airway remodeling and to assess the preventive effects of anti-angiogenic drugs on this process in a murine AR model.
MATERIALS AND METHODS
Animals
Four week-old female BALB/c mice (20-30 g) were used in all experiments. The study protocol followed the principles for laboratory animal research, as outlined in the Animal Welfare Act and Department of Health, Education, and Welfare guidelines for the experimental use of animals (National Institutes of Health), and was approved by our institution's animal subjects committee.
Sensitization, anti-angiogenic drug delivery, and allergen challenge
Forty-eight mice were divided into the following six groups: negative control mice challenged with phosphate-buffered saline (PBS; group A), mice challenged with ovalbumin (OVA, Grade V; Sigma, St. Louis, MO, USA; group B), control mice treated with dimethyl sulfoxide (DMSO; Sigma) prior to intranasal OVA challenge (group C), mice treated with SU1498 dissolved in DMSO prior to intranasal OVA challenge (group D1), mice treated with AG1296 dissolved in DMSO prior to intranasal OVA challenge (group D2), and mice treated with both SU1498 and AG1296 dissolved in DMSO prior to intranasal OVA challenge (group D3).
The procedure for allergen sensitization and challenge was performed as previously described and is summarized in Fig. 1.6 Briefly, mice were sensitized on days 0, 7, 14, and 21 by intraperitoneal injection with 300 µL PBS alone, or PBS with 25 µg OVA plus 1 mg aluminum hydroxide (Sigma). Starting on day 28, air with 2% aerosolized OVA was administered for 30 minutes daily for 7 days using a nebulizer (PulmoAide, Somerset, PA, USA). Aerosolized OVA (particle size, 0.5-5.0 µm) was generated by passing the solution through a closed 8,800 cm3 (20×22×20 cm) acrylic chamber. Mice received a single intraperitoneal injection (9 mg/kg) of SU1498 (#T-2710; Calbiochem, La Jolla, CA, USA; group D1), AG1296 (Cayman Chemical, Ann Arbour, MI, USA; group D2), or SU1498 plus AG1296 (group D3) dissolved in DMSO 4 hours prior to each aerosolized OVA exposure.19 Group C received an intraperitoneal injection of DSMO alone. Then 24 hours after the final OVA challenge, half of the mice were sacrificed (0 month group). The remaining mice received the 2% OVA aerosol by inhalation twice a week for the next 3 months (3 month group). Group A received PBS alone by inhalation. Each mouse was sacrificed 24 hours after the final OVA challenge, and their nasal tissues were harvested for subsequent analysis.
Evaluation of allergic responses
Nasal symptom scores. Prior to sacrifice, mice were subjected to intranasal provocation with 200 µg OVA, and the frequency of sneezing and nose scratching was monitored for 15 minutes to evaluate early allergic responses.
Measurement of total IgE and OVA-specific IgE. Serum levels of total IgE and OVA-specific IgE were measured by solid-phase enzyme-linked immunosorbent assay (ELISA). Serum samples were collected 24 hours after the final nasal OVA challenge, and 10 µg/mL of OVA was used to coat a microtiterplate. Bound immunoglobulin isotypes were detected with specific secondary antibody (biotin-conjugated rat antimouse IgE mAb was purchased from BD Pharmingen, San Jose, CA). Total serum IgE was measured by standard ELISA using antimouse IgE capture mAb (BD Pharmingen) and detection antibody was measured as described for the OVA-specific IgE ELISA.
Measurement of cytokines in splenocyte culture supernatants. Spleens were harvested 24 hours after the final intranasal challenge. Single-cell suspensions were plated on 96-well plates at a final concentration of 5×106 cells/well in RPMI-1640 containing 10% fetal bovine serum and penicillin/streptomycin. The cells were stimulated with OVA for 72 hours. Supernatants were collected and stored at -70℃ until analysis.
IL-4 and TGF-β were measured in OVA-stimulated splenocyte culture supernatants using commercially available ELISA kits (R&D Systems, Inc., Minneapolis, MN, USA). After measuring the optical density at 450 nm, the concentrations of IL-4 and TGF-β were calculated by interpolation from a standard curve and expressed as ng/mL.
Histological assessment
Mice were sacrificed 24 hours after the final OVA challenge, and nasal tissues were obtained for analysis. Two sections of the nasal septum, 4 µm apart, were made 5 mm posterior to the nasal vestibule and used for histological assessment.
Eosinophil counts. The nasal septum sections were stained with hematoxylin and eosin to assess inflammatory cell infiltration. Eosinophils were morphologically defined by the presence of eosinophilic granules in the cytoplasm and a 2-lobed nucleus, and they were counted under a microscope at ×400 magnification.
Periodic acid-Schiff (PAS) staining for goblet cell hyperplasia. PAS staining was performed on the nasal septal mucosa sections to visualize the development of goblet cell hyperplasia. The results are presented as the number of goblet cells per 100 µm nasal septal mucosa.
Masson's trichrome staining. Masson's trichrome staining was used to reveal the subepithelial deposition of collagen in the nasal mucosa. Positive trichrome-stained areas were quantified using ImageJ software (National Institutes of Health, http://rsbweb.nih.gov/ij/) to assess the degree of subepithelial fibrosis. Positive-stained areas for each group of mice were reported as percentages of positive-stained area/whole area. All slides were read by a single pathologist who was blinded to the experimental conditions.
Immunohistochemistry
Nasal septal musosa sections were stained immunohistochemically to evaluate the expression of MMP-9 and TIMP-1. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide in methanol. Nonspecific antigenic sites were blocked with 0.3% Triton X-100 plus 10% normal donkey serum in PBS at 37℃ for 2 hours. Then the sections were incubated overnight at 4℃ with anti-mouse mAb against MMP-9 or TIMP-1 (R&D Systems) at a dilution of 1:50. Isotype controls were also included. Next, the sections were incubated in the dark for 1 hours at room temperature with an anti-rabbit secondary antibody conjugated to AlexaFluor 594 (Invitrogen, Carlsbad, CA, USA) diluted in 0.01% Triton X-100 plus 1% normal donkey serum in PBS at a final dilution of 1:500. Finally, the sections were counterstained with 5 µg/mL 4',6-diamidino-2-phenylindole (DAPI) to stain cell nuclei and imaged using a confocal microscope (LSM510 META; Carl Zeiss, Gottingen, Germany).
RESULTS
Effect of anti-angiogenic drug treatment on allergy symptoms and production of IgE
The frequency of nasal rubbing and sneezing by the positive control and DMSO control mice at 3 months scored 7.8±1.3 and 6.8±1.0, respectively, which was significantly higher than that of the negative control mice (0.8±1.0, P=0.000). The symptom scores of mice treated with anti-angiogenic drugs (group D1=6.0±4.5, group D2=4.8±1.1, and group D3=5.8±1.7) did not significantly differ from that of positive control mice at 3 months (P=0.149).
All OVA treated mice had significantly higher total IgE, as well as OVA-specific IgE, than negative control mice at both 0 and 3 months (P<0.001). However, no differences were observed, comparing groups B, C, D1, and D2 with group D3, in levels of total IgE (P=0.861 at 0 months and P=0.605 at 3 months) or OVA-specific IgE (P=0.489 at 0 months and P=0.093 at 3 months).
Effect of anti-angiogenic drug treatment on eosinophilic inflammation
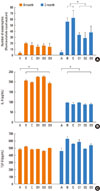
Eosinophil counts in the nasal mucosa are shown in Fig. 2A. At 0 months, eosinophil counts were similar in all OVA-treated groups of mice (P=0.512). At 3 months, the mean eosinophil counts in groups B and C were 55.5±6.9 and 62.3±12.5, respectively. In the drug-treated mice, eosinophil counts averaged 33.2±5.0 (group D1), 28.2±10.3 (group D2), and 37.6±11.0 (group D3), which were all significantly reduced compared to groups B and C (P<0.001).
Effect of anti-angiogenic drug treatment on the production of inflammatory cytokines
IL-4 was significantly elevated in OVA-treated mice (groups B, C, D1, D2, and D3), compared to the negative control mice (group A), as measured in splenocyte culture supernatants at both 0 (P<0.001) and 3 (P<0.001) months (Fig. 2B). However, neither SU1498 nor AG1296 treatment affected OVA-induced IL-4 production by splenocytes (P=0.230 comparing groups B, C, D1, D2, and D3 with group A at 0 months and P=0.246 for the same comparison at 3 months). There were no significant differences in TGF-β production by splenocytes from OVA-treated versus control mice (P=0.116 at 0 months and P=0.093 at 3 months) (Fig. 2C).
Effect of anti-angiogenic drug treatment on nasal airway remodeling
All mice showed minimal subepithelial trichrome staining in their nasal septal mucosa at 0 months (Fig. 3A). Areas that stained positive for trichrome were markedly increased in the OVA-treated mice in groups B and C at 3 months, but were significantly less in the OVA-treated mice that also received anti-angiogenic drugs (groups D1, D2, and D3; P<0.001) (Fig. 3B). A quantitative summary of positive-stained areas for each group is shown in Fig. 3C.
Goblet cell hyperplasia
Differences in goblet cell counts are summarized in Fig. 4. At 0 months, goblet cell numbers were similar in all groups of mice. At 3 months, all OVA-treated mice (groups B, C, D1, D2, and D3) had significantly more goblet cells than negative control mice (group A; P<0.001). No significant differences were found between groups B, C, D1, D2, and D3 (P=0.625).
Effect of anti-angiogenic drug treatment on MMP-9 and TIMP-1 expression
At 0 months, MMP-9 and TIMP-1 expression was sparse in the nasal mucosa of mice from all groups (Fig. 5A). At 3 months, higher MMP-9 and TIMP-1 expression was found in the basal and subepithelial layers of nasal tissue. OVA-treated mice (groups B and C) had more MMP-9 and TIMP-1 expression than PBS-treated mice (group A). Drug-treated mice (groups D1, D2, and D3) had lower MMP-9 and TIMP-1 expression than OVA-treated (positive control) mice (Fig. 5B). No significant differences were found between the various drug treatment groups.
A quantitative assessment of MMP-9 and TIMP-1 expression in the nasal septum of the different treatment groups is summarized in Figs. 5C and 6. At 0 months, all groups of mice showed similar levels of MMP-9 and TIMP-1 expression. At 3 months, OVA-treated mice (groups B, C, D1, D2, and D3) had significantly higher levels of MMP-9 and TIMP-1 than negative control mice (P<0.001). Mice treated with the anti-angiogenic drugs (groups D1, D2, and D3) showed significantly lower MMP-9 and TIMP-1 expression compared to groups B and C (P<0.001 for MMP-1 staining and P<0.001 for TIMP-1 staining). The ratio of MMP-9 to TIMP-1 expression was calculated for each group of mice at both time points, but no statistically significant differences were observed between the groups.
DISCUSSION
In this study, we showed that OVA challenge of AR mice for 3 months induced nasal airway remodeling, as demonstrated by increased subepithelial fibrosis, goblet cell numbers, and MMP-9/TIMP-1 expression in the nasal septal mucosa. Importantly, these airway-remodeling processes, with the exception of goblet cell hyperplasia, were prevented with long-term treatment with anti-angiogenic drugs.
Angiogenesis is a complex and dynamic process that is characterized by a coordinated sequence of cellular interactions.20 VEGF is known to regulate the proliferation, migration, and survival of vascular endothelial cells. In addition to acting as an angiogenic factor, VEGF has been identified as a survival factor for endothelial cells and newly formed blood vessels.21 Blood vessels undergoing maturation, on the other hand, are not dependent on VEGF.22 Instead, they are supported by pericytes, which provide stability while PDGF mediates their maturation.22 Consequently, inhibition of multiple angiogenic pathways, for example VEGF and PDGF, may be more effective at blocking angiogenesis than targeting one particular angiogenic factor. Targeting multiple angiogenic signaling pathways by polyvalent tyrosine kinase inhibitors has been investigated as a promising strategy to interfere with angiogenesis.20,23 In fact, previous reports have documented that the combined inhibition of VEGF and PDGF signaling effectively inhibits neovascularization.20,22,23
SU1498 ((E)-3-(3, 5-Diisopropyl-4-hydroxyphenyl)-2-[(3-phenyl-n-propyl) aminocarbonyl] acrylonitrile) is a potent and selective inhibitor of Flk-1 kinase, a VEGF receptor kinase. AG1296 (6, 7-Dimethoxy-3-phenylquinoxaline) inhibits PDGF receptor kinase, which blocks signaling through human PDGF α-receptors and β-receptors. In this study, we investigated the effects of SU1498 and AG1296 alone, or in combination, on airway remodeling in an AR mouse model. These receptor inhibitors are known to decrease VEGF and PDGF production, respectively, by inhibiting the migration and inflammatory responses of angiogenic factor-producing cells or blocking the autoinductive VEGF or PDGF pathway.18
MMP-9 is a zinc- and calcium-dependent, substrate-specific endopeptidase, and is the major proteinase involved in the turnover of the extracellular matrix and basement membrane proteins in respiratory epithelium. TIMPs are a family of secreted proteins that selectively, but reversibly, inhibit metalloproteinases.24 Therefore, MMP-9 and TIMP-1 are considered to be involved in the tissue remodeling process induced by inflammation. The increased expression of MMP-9 and TIMP-1 that we detected in the nasal septal mucosa after long-term OVA challenge is consistent with previous reports.25,26
A positive correlation between VEGF and MMP-9 levels in asthmatic patients has been documented.18 Furthermore, in a previous study, VEGF receptor inhibitors were effective at reducing MMP-9 expression, and reversing all pathophysiologic signs, in a murine asthma model,18 consistent with our findings. We also determined that infiltrating eosinophils were significantly reduced in the AR mice treated with SU1498 or AG1296. Eosinophils are the main effector cells of allergic inflammation and an important source of VEGF, along with other inflammatory and structural cells in inflamed tissue.27 Eosinophils also produce several other molecules, such as fibroblast growth factor-2, MMP-9, TIMP-1, IL-13, and IL-17, all of which are implicated in tissue remodeling processes.28
One plausible mechanism of nasal airway remodeling is that VEGF increases vascular permeability, causing leakage of plasma proteins and inflammatory cells, including eosinophils, into the extravascular space. This relocation of eosinophils, or other inflammatory cells, may cause the upregulation of MMP-9 and TIMP-1. Inhibition of plasma leakage and the subsequent migration of eosinophils by administering SU1498 and/or AG1296 may therefore reduce MMP-9 and TIMP-1 production. Enhanced eosinophilic inflammation in the nasal mucosa of human AR patients in response to VEGF secretion has been demonstrated.29 Our data using AR mice suggest that MMP-9 is generated in response to long-term allergen challenge and may be produced by local airway inflammatory cells, likely eosinophils.
The PDGF family of proteins includes important mitogens and chemoattractants for monocytes, and PDGF-B, in particular, is known to induce a variety of MMPs, including MMP-2 and MMP-9.30 A recent study revealed that PDGF facilitates the migration of airway smooth muscle cells by modifying the MMP-TIMP balance via the ERK pathway.31 PDGF is also an important activator of eosinophils in pulmonary inflammation associated with asthma.32
We observed goblet cell hyperplasia after long-term repetitive allergen challenge of AR mice. However, administration of anti-angiogenic drugs did not inhibit the hyperplasia process. Goblet cell hyperplasia typically occurs in response to various airway insults. Cytokines (e.g., interleukin-4, -9, and -13), bacterial products (e.g., lipopolysaccharide and lipoteichoic acid), proteinases (e.g., elastase and cathepsin G), and oxidants from T helper-2 (Th2) lymphocytes, bacteria, and neutrophils have all been shown to induce goblet cell hyperplasia.33 Therefore, the inhibition of neovascularization and inflammatory cell infiltration by administration of anti-angiogenic drugs may not be sufficient to suppress the hyperplasia process. For example, the production of IL-4 was not affected by the administration of anti-angiogenic drugs in our study. Th2 lymphocyte-derived cytokines, including IL-4, IL-9, and IL-13, are known to be highly effective in inducing goblet cell hyperplasia.34
In this study, neither eosinophil infiltration nor MMP-9/TIMP-1 expression was enhanced by the combined use of SU1498 and AG1296, as compared to injecting either drug alone. VEGF and PDGF are responsible for the initiation and maturation of neovascularization, respectively.20 If neovascularization itself were the leading cause of airway remodeling, synergistic effects using both drugs would be expected. Instead, our results suggest that increased vascular permeability followed by eosinophil infiltration may explain the increased expression of MMP-9/TIMP-1. Alternatively, the administrative dose of these drugs may have been too low to detect synergistic effects. In addition, we injected mice with a single solution containing both VEGF and PDGF inhibitors (group D3). We may have seen different results if we had administered the inhibitors separately, each in a discrete injection site.
Administration of anti-angiogenic drugs partially inhibited the expression of MMP-9 and TIMP-1, and also prevented subepithelial fibrosis in this study. MMP-9 and TIMP-1 are synthesized and secreted by connective tissue, comprising fibroblasts, endothelial cells, and inflammatory cells (including eosinophils).35 Thus, other sources of MMP-9 and TIMP-1 may have compensated for the drug-induced reduction, explaining our observation of only partial inhibition. Alternatively, the administrative dose of the anti-angiogenic drugs we used to reduce plasma leakage and migration of inflammatory cells may have been too low to completely prevent the remodeling process. Further studies analyzing the dose-response relationship will help to clarify this discrepancy.
In summary, the results of this study demonstrate that long-term and repetitive allergen challenge induces eosinophil infiltration, MMP-9/TIMP-1 release, and evidence of airway remodeling in the nasal cavity of AR mice. Although anti-angiogenic drugs (i.e., VEGF and PDGF receptor inhibitors) did not influence the early allergic response or inflammation, they were able to prevent the airway remodeling process during exposure to a long-term allergen challenge to the nasal airway. These results suggest that clinical use of angiogenic inhibitors may help treat hypertrophy of the nasal septum or turbinates as well as AR.




 XML Download
XML Download