

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Acute interstitial pneumonia (AIP), a rapidly progressive respiratory disease that affects patients with no pre-existing lung disease, is a lethal lung disease with a mortality rate of about 50%.1 Unclassified interstitial pneumonia with fibrosis, which is not classified as a disease entity under the classification of interstitial lung diseases in adults,1 shows clinical and radiological findings that are similar with those of AIP. However, the pathological findings of unclassified interstitial pneumonia are different from those of AIP. AIP is characterized by histological findings of diffuse alveolar damage (DAD) in the acute phase with subsequent fibrotic organization, whereas unclassified interstitial pneumonia with fibrosis is characterized histologically by centrilobular distribution of alveolar damage and bronchiolar destruction with bronchiolar obliteration. An increase in the incidence of unclassified interstitial pneumonia with fibrosis in Korean children was observed during March to June 2006.2 This sudden increase in reported cases might have been the result of clinicians' previous lack of awareness of the disease. Alternatively, it may indicate the occurrence of an epidemic.3 In this report, we describe two familial case series of unclassified interstitial pneumonia with fibrosis, which developed almost simultaneously in the spring. These involved a previously healthy father and son (Family 1), and a mother and two daughters (Family 2).
CASE REPORTS
Family 1, Case 1
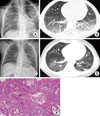
A previously healthy 2-year-old boy presented with a 1-month history of cough and dyspnea. He had had a mild cough for 1 week before a sudden deterioration in his condition. The dyspnea increased despite treatment, and he was transferred to the Department of Pediatrics at the Asan Medical Center for further management. On arrival, he was apyrexial and tachypneic (respiratory rate, 66/min). Mild subcostal retraction was observed, and coarse breath sounds without crackles were noted on auscultation. Arterial blood gas analysis (ABGA) showed mild hypoxemia. His white blood cell (WBC) count was 14,300/µL, with 55.8% neutrophils and 36.3% lymphocytes. Radiography and computed tomography (CT) of the chest revealed the presence of fine peribronchial ground-glass opacities in both lungs (Fig. 1A and 1B). Blood, bronchoalveolar lavage (BAL) fluid, and sputum cultures were negative for bacteria, viruses, and fungi. A lung biopsy performed on day 3 of admission showed the organizing phase of DAD distributed mainly in the centrilobular area, with destruction and obliteration of bronchioles by fibroblasts (Fig. 1E). The patient was administered intravenous corticosteroids (2 mg/kg/day), followed by oral prednisolone (which was gradually tapered), hydroxychloroquine, and oral cyclophosphamide. His condition gradually improved, although exercise intolerance persisted. At the 1-year follow-up, a repeat CT scan of the chest revealed a decrease in the extent of ground-glass opacities in the affected areas of both lungs (Fig. 1C and 1D).
Family 1, Case 2
A previously healthy 47-year-old non-smoking male patient presented with a 2-day history of cough, mucoid sputum, and chills. His son was hospitalized with the same symptoms at the same time. On physical examination, he was apyrexial, tachycardic, and tachypneic, and rales were audible over the left lung field on auscultation. Chest radiography showed diffuse hazy patches in the left lung, and a chest CT scan revealed the presence of multifocal consolidation and diffuse centrinodular ground-glass opacities in both lungs. Laboratory findings included a normal WBC count, an elevated erythrocyte sedimentation rate (ESR) of 42 mm/hr, an elevated C-reactive protein (CRP) level of 5.19 mg/dL, and a normal ABGA. Laboratory tests for collagen disease and vasculitis were negative. Blood, sputum, and BAL fluid cultures were negative for bacteria, viruses, and fungi. Lung biopsy, performed on day 11 of admission, showed histological features of fibroproliferative DAD with diffuse interstitial fibrosis and relative sparing of the peripheral lobular air spaces. The patient required mechanical ventilation for a total of 36 days. He was administered broad-spectrum antibiotics and high-dose corticosteroids. On discharge, he still experienced dyspnea on exertion and required intermittent oxygen at night and on exertion. At the 1-year follow-up, radiography and a CT scan revealed a decrease in the degree of bronchial dilatation and peribronchial consolidation; the patient still complained of dyspnea on exertion.
Family 2, Case 3
A previously healthy 35-month-old girl was referred to our medical center for severe dyspnea. Four weeks prior to admission, she had developed a mild cough, and her oral intake had become poor. Four days prior to admission, she had developed sudden onset dyspnea and subcutaneous emphysema. On admission, she presented with marked tachypnea (respiratory rate, 80/min) and chest retraction, and rales were audible in both lungs on auscultation. Radiography and a CT scan of the chest showed an extensive pneumomediastinum, a pneumothorax in the right hemithorax, and diffuse ground-glass opacities with consolidation in both lungs (Fig. 2A and 2B). Her total leukocyte count was 9,300/µL, with 66% neutrophils and 27% lymphocytes. Blood, sputum, and BAL cultures for bacteria and fungi were negative. Multiplex RT-PCR of nasopharyngeal aspirates revealed the presence of respiratory syncytial virus (RSV). She was admitted to the intensive care unit and treated with steroids, cyclophosphamide, hydroxychloroquine, and broad-spectrum antibiotics. Her response to treatment was poor, and her condition deteriorated rapidly. On the third day of admission, mechanical ventilation was required for impending respiratory failure. Repeated chest radiography revealed the presence of increased opacities in both lungs (Fig. 2C). She then developed refractory hypoxemic and hypercarbic respiratory failure with subsequent multiorgan dysfunction. Despite the administration of antifibrotics and supportive therapy, she died 70 days after admission. A postmortem lung biopsy revealed the features of the fibrotic phase of DAD with occasional hyaline membrane and type II pneumocyte hyperplasia (Fig. 2D).
Family 2, Case 4
A 30-year-old non-smoking woman, who was the mother of Cases 3 and 5, was admitted to the Department of Internal Medicine at the Asan Medical Center with a 1-month history of cough and a 10-day history of dyspnea on exertion. On physical examination, she was apyrexial and tachypneic (respiratory rate, 44/min), and crackles were audible over the right lung on auscultation. No hypoxemia was detected by ABGA. Her leukocyte count was 14,000/µL, with 72% neutrophils and 20% lymphocytes. The ESR was 38 mm/hr; and the CRP was 1.92 mg/dL. Laboratory tests for collagen vascular disease and vasculitis were negative. Blood, sputum, and BAL fluid cultures were negative for bacteria, viruses, and fungi. Pulmonary function tests showed a mild restrictive defect. Radiography and a CT scan of the chest revealed the presence of multifocal patchy consolidation with ground-glass opacities and tiny nodular lesions in both lungs. The patient was treated with steroid pulse therapy, followed by oral steroids, azathioprine, and antibiotics. The dyspnea gradually decreased, and she was discharged after 2 weeks of treatment. A repeat CT scan of the chest 12 days post-discharge showed an increase in the extent of ground-glass opacities and tiny nodules in both lungs. She was readmitted 14 days post-discharge with increased dyspnea. Cyclosporine was added to her existing treatment regimen of steroids and azathioprine. Transbronchial biopsy showed a nonspecific subacute lung injury pattern, including focal-organizing pneumonia and mild interstitial chronic inflammation. A follow-up chest CT scan 4 weeks after readmission revealed mild resolution of the abnormalities in both lungs. After discharge, her daily activities were limited by dyspnea, and she continued to experience dyspnea at rest.
Family 2, Case 5
A 17-month-old girl presented to the out-patient clinic at our medical center with a 30-day history of worsening cough. Her older sister (Case 3) had been admitted to the intensive care unit at the Asan Medical Center with respiratory failure 1 month previously, and her mother (Case 4) had had similar symptoms during the previous 45 days. On physical examination, the patient was apyrexial, tachycardic, and tachypneic (respiratory rate, 40/min). Her WBC count was 21,900/µL, with 48% neutrophils and 43% lymphocytes. Blood, sputum, and BAL fluid cultures were negative for bacteria, viruses and fungi. However, multiplex RT-PCR of nasopharyngeal aspirates revealed the presence of rhinovirus. Radiography and a CT scan of the chest showed the presence of centrilobular ground-glass opacities and septal thickening in both lungs. She was treated with steroids, cyclophosphamide, and hydroxychloroquine for 2 weeks. She showed significant clinical improvement, and repeat chest radiography 2 weeks after the initiation of treatment revealed that the ground-glass opacities, septal thickening, and consolidation had decreased. After discharge, she did not require oxygen, and her daily activities were not restricted, although she continued to experience dyspnea on exertion.
DISCUSSION
In this study, we described two series of familial cases of interstitial lung disease of unknown etiology, in a young male patient and his father, and two young girls and their mother. They presented with the same clinical symptoms, showed rapidly progressive respiratory failure of unknown cause, and had similar radiological and pathological features, including rapidly progressive lung fibrosis. As this disease has not been previously described, we named this disease entity 'unclassified interstitial pneumonia with fibrosis.' Previously, we reported this unclassified interstitial pneumonia with rapidly progressive pulmonary fibrosis as AIP.2,3
AIP is a potentially fatal respiratory disease of unknown etiology that presents in previously healthy individuals.4-8 Unclassified interstitial pneumonia with fibrosis, which is not classified as any of the disease entities in the classification of interstitial lung diseases in adults,1 shows clinical and radiological findings that are similar to those of AIP. However, these two diseases have different pathological findings. Unclassified interstitial pneumonia with fibrosis is characterized histologically by centrilobular distribution of alveolar damage and bronchiolar destruction with bronchiolar obliteration.
This unclassified interstitial pneumonia with fibrosis commonly presents with an acute onset of cough and/or dyspnea in a previously healthy individual, similar to AIP.4,8 Symptoms progress rapidly and may result in hypoxemia and cyanosis. Chest radiography in AIP reveals the presence of progressive airspace consolidation.6,9 Chest CT scans in AIP reveal extensive bilateral airspace consolidation and patchy or diffuse ground-glass attenuation.6,9 In this report, we present two familial series of cases with unclassified interstitial pneumonia with fibrosis. The chest CT scans showed a pattern of consolidation predominantly in the lower lungs, with subpleural sparing in the early phase. In the late phase, chest CT scans showed diffuse centrilobular nodular opacity. Lung function tests revealed restrictive patterns in cases 2 and 4. Lung function tests were impossible in the other young cases because of non-cooperation.
Although interstitial lung disease has been considered idiopathic, factors such as medications, radiation, infections, and environmental and occupational agents have been implicated in its development.6-8 Within each of the familial case series presented, the patients shared the same environment and experienced an almost simultaneous onset of similar clinical symptoms (Table). The lung biopsy specimens and radiological findings in these case series showed a similar pattern across affected family members, suggesting that a common environmental insult or viral infection might have been a causal or triggering factor for the development of interstitial pneumonia with lung fibrosis in these patients.
The association between interstitial pneumonia and genetic mutations in a family raised the question of whether genetic mutation could be responsible for the simultaneous development of this disease in family members of different ages. Although genetic susceptibility cannot be ruled out in these familial cases, the simultaneous development of interstitial pneumonia implies the involvement of exposure to common environmental factors or the presence of infection in each family member. Because all family members reside in an urban region with no history of contact with animals or dust, it is suggested that interstitial pneumonia may be associated with viral infections; however, more investigation is needed in the future.
A nationwide pediatric surveillance study performed in 2006 and then again in 2008 demonstrated a geographical and seasonal pattern to the increased incidence of unclassified interstitial pneumonia with fibrosis in Korea.3 Between February and June in both 2006 and 2008, an increase in the incidence of unclassified interstitial pneumonia with fibrosis was initially observed in the southern regions of the country, and this progressed gradually toward the northern regions.2,5 The spring timing of this interstitial pneumonia, its characteristic pattern of spread from the south to north, and the detection of various viruses (i.e., human corona virus, cytomegalovirus, and parainfluenza virus), when considered together with the findings of the present familial case series, provide evidence that viral infection may be associated with the development of unclassified interstitial pneumonia with fibrosis in Korea. Altered or exaggerated host defense mechanisms, or repair processes triggered by a viral infection, may be responsible for the lung damage and fibrosis observed in these patients.
Early lung biopsy is indicated in cases of suspected interstitial pneumonia. Within 3 weeks of symptom onset, fibroblastic proliferation is evident on histological examination, and this manifests clinically as rapidly progressing respiratory failure.6,7 In Case 1 of the present report, although histological examination revealed fibroproliferation, this patient experienced a better outcome than the other four cases. This better outcome indicates that rapid diagnosis and early therapeutic intervention may inhibit disease progression and increase survival in patients with unclassified interstitial pneumonia with fibrosis.
Although there is no proven effective therapy for unclassified interstitial pneumonia with fibrosis, recommended treatments include supportive care and immunosuppressive therapies such as corticosteroids, cyclophosphamide, and vincristine.7,10 These treatments were found to be most effective during the early proliferative phase, i.e., before the development of fibrosis in the late fibrotic phase.9 Early treatment with corticosteroids, cyclophosphamide, and hydroxychloroquine, a combination designed to inhibit fibrogenesis, might have contributed to the more favorable outcome in Case 1 of the present series. In addition, the presence of a pneumothorax and/or a pneumomediastinum on chest radiography (case 3) and the severity of the respiratory difficulty, both reflecting disease severity, may predict poor response to treatment.
In summary, this report describes two series of familial cases of unclassified interstitial pneumonia with fibrosis, presenting rapidly progressing respiratory failure. The occurrence of unclassified interstitial pneumonia with fibrosis only in spring and its familial pattern suggest that the development of this disease may be related to exposure to viruses or to common inhaled environmental agents. Further studies are needed to better understand the etiology and pathophysiology of unclassified interstitial pneumonia with fibrosis. As this disease has a poor prognosis, early diagnosis with lung biopsy and radiological examination, and early initiation of immunosuppressive therapy are critical for improving the outcome.


 XML Download
XML Download