

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Drug-induced liver injury (DILI) is an unpredictable adverse drug reaction that affects only a small subset of treated patients. As a result, DILI is scarcely detected during pre-approval clinical trials, and therefore remains the single leading cause of drug withdrawal from the market.1 Liver injury has been linked to nearly 1,000 drugs and DILI is the most common cause of acute liver failure in the US, where it accounts for more than half of all acute liver failures.2,3 The mechanism underlying DILI is thought to be two-staged and likely involves non-immune and immune-mediated stages. Liver injury is initiated by the direct hepatotoxic effects of a drug or its reactive metabolites, and subsequently, the resulting parenchymal cell injury induces activation of innate and/or adaptive immune cells. This immune cell activation in turn produces proinflammatory and hepatotoxic mediators and initiates immune reactions against drug-associated antigens.1,4 Many studies suggest that DILI results from the production of reactive metabolites that accumulate within hepatocytes at levels that exceed some critical threshold.1,5,6
We hypothesized that there could be a common susceptibility gene for DILI that acts during the initiative non-immune-mediated stage. Based on the results of a microarray analysis from previous studies on toxic hepatitis in animal models,7,8 we selected the thioredoxin reductase 1 gene (TXNRD1) as a potential marker of DILI and performed this genetic association study. TXNRD1 has been found to be commonly dysregulated in previous studies using DNA microarray analyses of toxic hepatitis induced by various compounds in animal models.7,8 TXNRD1 is a NADPH-dependent enzyme and is known to be related to oxidative stress and the regulation of cell growth and transformation.
MATERIALS AND METHODS
Study subjects and phenotype definitions
Records from 118 patients with DILI were extracted from the database of the Adverse Drug Reaction Research Group in Korea, members of which are drawn from 6 tertiary hospitals (Seoul National University Hospital, Hanyang University Hospital, Ajou University Hospital, Dankook University Hospital, Seoul National University Bundang Hospital, and Eulji University Hospital). DILI was defined as drug-induced acute hepatocellular injury with an aspartate aminotransferase level≥upper limit of normal (ULN) and an (aspartate aminotransferase/ULN)/(alkaline phosphatase/ULN) ratio≥5.10 No patients showed any other organ involvement other than liver involvement, except peripheral eosinophilia. The time between the initiation of the causative drug and symptom onset was ≤14 days. Causality between hepatitis and each drug was assessed using the Naranjo algorithm, which is used to assess the likelihood that a change in clinical status is the result of an adverse drug reaction rather than the result of other factors such as the progression of an underlying disease. The Naranjo algorithm uses a scoring system that is related to the possibility of underlying disease exacerbation, previous reports of drug adverse reaction, timing of onset, results of de-challenge or re-challenge, and any objective evidence of drug adverse reaction.11,12 One hundred and twenty drug-matched controls were recruited from the above-mentioned hospitals. All control patients had been prescribed the same drugs but had not developed liver injury. All subjects enrolled in this study provided written informed consent, and the study protocol was approved by the Institutional Review Boards at each of the above-mentioned hospitals.
Gene and single nucleotide polymorphism selection and genotyping
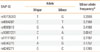
After reviewing the results of previous microarray analysis studies conducted on toxic hepatitis animal models, we selected TXNRD1 as a candidate gene.7,8 Based on Japanese data in HapMap (Japanese genetic constitutions are similar to those of Koreans), 7 tagging SNPs (rs10735393, rs4964287, rs4595619, rs10861201, rs11111997, rs4246270, and rs4246271) on TXNRD1 (minor allele frequency≥5% and linkage disequilibrium coefficient r2≥0.8) were selected (Table 1; Figure). Haplotypes and their frequencies were estimated using an expectation-maximization algorithm using Haploview 3.32 (available online at http://www.broad.mit.edu/mpg/haploview). Inferred haplotype blocks are shown in Figure. Scoring was performed using the high-throughput single base-pair extension method (SNP-ITTM assay) with an SNPstream25K system customized to automatically genotype DNA samples in 384-well plates, and to provide a colorimetric readout (Orchid Biosciences, Princeton, NJ, USA), as described previously.13
Statistical analysis
An association analysis was performed for each SNP using an allelic model (A versus B, where A is the major allele and B the minor allele [for haplotype analysis, a haplotype vs. the other haplotypes]), which assessed the elevated risk of disease development in individuals carrying a risk allele (or haplotype). P values and odds ratios (ORs) were determined by logistic regression analysis and controlled for age and sex. Compliance with Hardy-Weinberg equilibrium was determined using 2×2 tests. All statistical analyses were performed using SAS 9.1 software (SAS Institute Inc., Cary, NC, USA), and P<0.05 was regarded as statistically significant.
RESULTS
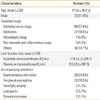
The mean age of the 118 cases was 46.9±18.3 years (mean±standard deviation [SD]) and 72 (61.0%) were male (Table 2). The mean age of the 120 controls was 49.8±11.3 years (mean±SD) and 95 (79.1%) were male. Neither age nor sex was significantly different in cases and controls. Causative drugs were antituberculosis drugs (n=68, 57.6%), antibiotics (n=22, 18.6%), antiepileptic drugs (n=7, 5.9%), non-steroidal anti-inflammatory drugs (n=5, 4.2%), and others (n=16, 13.7%).
We failed to find differences between the frequencies of any of the 7 SNPs in the cases and controls after Bonferroni correction (Table 3). However, we did find a significant association between the TTA haplotype (composed of rs10735393, rs4964287, and rs4595619) and DILI using an allele model (OR, 1.79; 95% confidence interval, 1.18-2.73; P=0.008; Bonferroni corrected P=0.024).
DISCUSSION
Two major mechanisms have been proposed to be responsible for DILI: the intrinsic hepatotoxicity of a particular drug or its metabolites and an immune-mediated reaction leading to hepatic inflammation and injury. Nevertheless, DILI is suspected to be initiated by liver damage in both mechanisms, which compromises the immunologically privileged status of the liver.14 As an example of a complex-trait disease, many genetic variants have been found for DILI that are associated with various processes, such as bioactivation/transport pathways, detoxifying enzymes, and transporters.15-20 Most of these genetic variants suggest that DILI results from the production of reactive metabolites that accumulate within hepatocytes to levels that exceed certain critical thresholds. This reactive metabolite hypothesis could explain individual susceptibilities to DILI related to genotype, poor metabolism, or a reduced capacity to resist or recover from injury.5,6 Furthermore, because liver damage is a common initial step of DILI caused by different drugs, there could be a common genetic basis to DILI susceptibility.
TXNRD1 is a NADPH-dependent enzyme that catalyzes the reduction of thioredoxin.9 TXNRD1 is known to play a role in cell growth and transformation and to protect cells against injury by oxidants. In a previous DNA microarray analysis of toxic hepatitis in animals (induced by several different hepatotoxic compounds), TXNRD1 was found to be commonly dysregulated under conditions of acute hepatotoxicity.7,8 Minami et al.7 sought to determine whether a relationship exists between the gene expression profiles and hepatotoxic phenotypes of 5 hepatotoxic chemicals in rats (acetaminophen, bromobenzene, carbon tetrachloride, dimethylnitrosamine, and thioacetamide), and found that TXNRD1 was upregulated in rats treated with these chemicals. Similarly, the expression of TXNRD1 was elevated in liver tissues after treatment with 6 hepatotoxic compounds (tetracycline, carbon tetrachloride, 1-naphthylisothiocyanate, erythromycin estolate, acetaminophen, or chloroform), but not after administration of non-hepatotoxic compounds.8 TXNRD1 is an enzyme with broad substrate specificity related to oxidant challenge, and thus, seems to be a good candidate common marker of DILI. However, despite recent enthusiasm, it seems that genetic variations in TXNRD1 do not entirely explain DILI. As has been suggested by other investigators, DILI appears to be caused by a combination of several genetic variations rather than a single mutation.16 Nevertheless, our results encourage us to consider that common genetic variations contribute to DILI development by various drugs.
Among the seven targeted SNPs, rs10735393 is located in the promoter region, rs4964287 is a synonymous SNP in the coding region, and the other SNPs are located in the intron region. There have been no functional studies related to these seven SNPs. However, as mentioned above, TXNRD1 has been found to be commonly dysregulated in previous studies using DNA microarray analyses in animal models of toxic hepatitis induced by various compounds.7,8
Several important limitations of the present study should be mentioned. First, different causative drugs related to DILI were examined in this study; therefore, the study population for each drug was too small to establish a common pathway for DILI. Second, the cases enrolled in the present study had comorbidities in addition to DILI, and this could have acted as a confounding factor. However, a variety of comorbidities and comparisons with healthy controls might dilute these confounding effects. In addition, the comparability of the case and control subjects was not fully investigated because the definition of control subjects totally depended on self-reported questionnaires. Furthermore, we could not completely remove the risk that spurious interactions might have been identified due to the relatively small size of the study population. Accordingly, we suggest that our results be confirmed by a larger study.
In summary, the present study shows that genetic variations in TXNRD1 significantly increase the likelihood of DILI. Although DILI is a rare and complex-trait disease, we believe that genetic variations in TXNRD1 could be a common marker of susceptibility for DILI by various drugs, although a larger confirmative study is needed.


 XML Download
XML Download