

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Asthma is a genetically complex disease that is associated with the familiar segregation of atopy and increased levels of total serum IgE.1 Asthma and atopy are also closely associated with increased bronchial hyper-reactivity and elevated blood eosinophil count.2,3 These intermediate phenotypes are highly heritable and the subject of much asthma genetics research. The occurrence of patients with an asthma cluster in their family indicates that a genetic component is likely operating. Twin studies represent a useful first step to determine whether a given trait or disease has a measurable genetic component. In a large twin study with 7,000 same-sex twins born between 1886 and 1925, the concordance rate for self-reported asthma in monozygotic twin pairs was 19%, which is four times higher than the 4.8% rate in dizygotic twins.4 This heritability must be determined by genetic factors. However, the prevalence of childhood and adult-onset asthma has increased dramatically during the last two or three decades in both developed and developing countries.5 These result indicate that epigenetic factors may play an important role in increased asthma prevalence. Genome-wide linkage studies, biologically plausible candidate gene approaches, and genome-wide association scans (GWAS) have been performed over the past 20 years to search for the genetic background of asthma.
WHOLE-GENOME LINKAGE, POSITIONAL CLONING, AND FINE MAPPING STUDIES
Linkage-based methods have been used for individual families in which members are highly affected by the disease. These studies have attempted to demonstrate a linkage between disease occurrence and genetic markers in a chromosomal region. Linkage analysis is performed to determine the chromosome location of a susceptibility gene or genes by demonstrating co-segregation of the trait or disease with known genetic markers, which are usually polymorphic DNA markers. Two general linkage analysis approaches have been used: 1) genome-wide and 2) candidate region searches of the genome in which candidate genes have already been mapped. These approaches have been successfully used to map and clone genes causing monogenic disorders with simple Mendelian inheritance such as cystic fibrosis.6 They have also been used over the past 20 years by several groups to isolate susceptibility loci for asthma.7,8 Whole-genome or candidate chromosome scans using microsatellite markers and a positional cloning approach has led to the discovery of several genes for asthma and related phenotypes (Table 1). These results have increased our understanding of the genetic background of asthma and its subphenotypes. Using whole-genome linkage analysis, positional cloning, and case control studies, at least five asthma genes including disintegrin and metalloprotease 33 (ADAM33) 20p13,9 dipeptidylpeptidase 10 2q14.1 (DPP10),10 plant homeodomain zinc finger protein 11 13q14.2,11 G protein-coupled receptor for asthma susceptibility 7p15-p14,12 and prostaglandin D2 receptor 14q2413 have been identified as strong genetic variants for asthma (Table 1). However, replication studies including the British 1958 Birth Cohort study of 7,703 adults revealed that small increases in asthma risk were identified only with DPP10 and ADAM33.9,10 Based on recent replication results, it has been concluded that applying linkage analyses to multi-factorial complex diseases is less successful than had been previously thought. Thus, the growing recognition of the limitations of linkage analysis for investigating a complex human disease has shifted emphasis away from linkage analysis and microsatellite markers toward single-nucleotide polymorphism (SNP) genotyping and different analytical strategies based on association and haplotype analyses.14
CANDIDATE GENE ASSOCIATION STUDIES USING SNPS
During the past decade, genetic and genomic molecular technologies have been rapidly developed. Generating SNP maps from high-throughput sequencing projects has promoted the discovery of genes related to asthma. There are several potential advantages of using SNPs to investigate the genetic determinants of a complex human disease such as asthma. SNPs are plentiful throughout the human genome, as they are found in exons, introns, promoters, enhancers, and intergenic regions, allowing them to be used as markers in dense positional cloning investigations. Additionally, groups of adjacent SNPs exhibit linkage disequilibrium and haplotypic diversity patterns that could be used to enhance gene mapping. More than 300 genes have SNPs associated with asthma and allergy subtypes based on the NCBI website (http://www.ncbi.nlm.nih.gov). We have also genotyped 2,800 SNPs on 180 genes whose functions are related to asthma and found that SNPs on 19 genes are associated with the risk of asthma and its traits (Fig. 1).15-30 Interestingly, almost all SNPs have an odds ratio (OR) <2.0, indicating that the candidate gene approach provides genetic variant information regarding the production of significant increases in asthma risk; however, their contribution to the development of asthma may be smaller than expected.
GWAS
With the development of SNP sequencing technologies, the international Hap-Map Consortium has revealed nearly 4 million SNPs on the whole chromosome and demonstrated that individual SNPs predict adjacent SNPs, suggesting that genotyping 500,000 SNPs may allow for a nearly complete survey of all common genetic variability.31 Based on this concept, whole-genome SNP genotyping arrays have been developed and applied over the past 5 years to research the genetic background behind multifactorial complex diseases. This approach is a hypothesis-free test of the association between phenotypes and SNPs across the genome and diseases, usually involving 500,000 markers that are reasonably polymorphic and are spread fairly evenly across the genome. As of June 2010, 904 statistically significant GWAS have been published for 165 traits of common allele complex diseases (NHGRI catalog at (http://www.genome.gov/gwastudies/). A GWAS on asthma has been published from 2007 until now (Table 2).21,32-38 Among the nine studies, seven consider Caucasians, one study considers Mexicans, and two studies consider Asians, including Koreans. Eight studies focused on asthma risk, and the remaining one focused on occupational asthma. The first GWAS was created in England in 2007.32 They applied a 30 kb SNP chip to the DNA from 994 patients with childhood-onset asthma and 1,243 non-asthmatics, using family and case-referent panels. As a result, a strong signal was found at chromosomes 17q and 2. The SNPs associated with childhood asthma were consistently and strongly associated (P=10-22) with transcript levels of ORMDL3, a member of a gene family that encodes transmembrane proteins anchored in the endoplasmic reticulum.32 The second GWAS analyzed sequence variants affecting eosinophil counts in the blood of 9,392 Icelanders. The most significant 10 SNPs were further studied in 12,118 Europeans and 5,212 east Asians, including Koreans. SNPS (rs1420101) at IL1RL1 and at 2q12 were strongly associated with asthma (P=5.5×10-12) in a collection of ten different populations including 7,996 cases and 44,890 controls. However, the ORs were <1.5. Very recently, a large-scale, consortium-based GWAS of asthma with 10,365 asthmatic patients and 16,110 controls confirmed the association of the previously defined SNPs, including ORMDL3 and IL1RL1.38 However, the ORs were within the range of 0.5 to 1.5. We calculated population attributable risk fractions of these SNPs in asthma and its traits,39 and they were between 3.9-24% (Table 2).
IDENTIFYING THE MISSING HERITABILITY OF ASTHMA
GWASs have identified hundreds of genetic variants associated with complex human diseases and traits and have provided deep insights into their genetic makeup. In the case of age-related macular degeneration, proportions of heritability explained by five SNPs are responsible for 50% of the risk. Thirty-two loci for Crohn's disease explain 20% of the heritability (Table 3). However, most variants identified to date confer a relatively small increase in risk and explain only a small proportion of heritability. Thus, this leads to the question of how the missing heritability can be explained.40 In the GWAS results, the population attributable risk fractions of these SNPs in asthma and its traits ranged between 3.9-24% (Table 2). The population attributable risk fraction (AF) of 20% indicates the relatively strong impact of genetic variants on the development of asthma. In studies including patients with asthma, rs7216389 on ORMDL3 has a 21.8% of AF, whereas the SNPs on the other gene have an AF of <12%. These results suggest that SNPs discovered even by several GWASs only have a limited ability to explain genetic effects for the development of asthma.
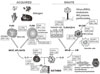
Manolio et al.40 regarded the factors related to the limitations of GWAS as an imprecise disease phenotype, use of control groups of questionable comparability, and inconsideration of environmental contributors. To overcome the missing heritability, the overall asthma strategy should be classified into specific phenotypes, and environmental factors should be introduced into the analysis of these subgroups. From a schematic view, the pathogenic mechanism of asthma can be divided into two major pathways. One is an acquired immune response pathway, and the other is an innate pathway (Fig. 1). In the former, Th2 cells, mast cells, and eosinophils participate in a antigen specific IgE- and Th2 cytokine-dependent manner. This process usually starts at a young age. In the latter, macrophages, dendritic cells, epithelial cells, and neutrophils are involved in IgE-independent adult-onset asthma. Thus, patients with asthma can be stratified into several subphenotypes (Table 4).
Based on the triggering environmental factors, asthma can be subgrouped into IgE-dependent allergic, aspirin-exacerbated respiratory disease, occupational asthma, exercise-induced asthma, and menstruation- or obesity-associated asthma. The inflammatory patterns observed from a sputum analysis reveal eosinophilic, neutrophilic, and pauci-graniulocytic types of asthma. Patients with asthma can be clinically and physiologically stratified into several subgroups. Thus, a genetic association study should be conducted on patients with asthma in well-defined subphenotypes.
IMPROVEMENT OF AF OF GENETIC VARIANTS ACCORDING TO THE SUBPHENOTYPES OF ASTHMA AND THE ENVIRONMENTS
The SNP on ORM1-like 3 was reanalyzed in Caucasians and Koreans according to the age of asthma onset (Table 5).41 When the subjects were stratified based on an age of 16 years, the statistical difference of rs 7216389 on 17q21 became more apparent in the age group <16 years in Caucasians and Koreans, whereas the statistical significance disappeared in the group >16 years. The ORs of the younger group ranged from 1.26 to 1.49, whereas that of the older group ranged from 0.87 to 1.11. AFs were 0.269 for early-onset asthma and 0.057 for late-onset asthma, indicating the necessity for stratifying asthma according to age, because the two types of asthma may have a different pathogenesis.
The impact of genetic variants on asthma may be enhanced when environmental factors are introduced. An interesting finding related to 17q21 variants was revealed after 1,511 subjects from 372 families were grouped by passive exposure to environmental tobacco smoke during early life.42 The ORMDL3 gene variant on rs8076131 showed a significant association with the risk of asthma in a family sample with offspring exposed to tobacco smoke (OR=2.5), but not in those who were not exposed to tobacco smoke (OR=1.38), indicating the importance of environment factors in genetic studies of asthma. One important and easily accessible environmental factor is occupational exposure to inducers or triggers.
A GWAS study was performed on a well-defined subphenotype of asthma in 84 Korean patients with toluene diisocyanate (TDI)-induced asthma and 263 unexposed healthy normal controls.21 Genetic catenin alpha 3, alpha-T catenin polymorphisms are significantly associated with the TDI-induced asthma phenotype (OR=5.84106 for rs10762058) and the AR increases to 24% (Table 2), indicating that the missing heritability of asthma can be compensated for by stratification into asthma sub-phenotypes and by introducing environmental factors into the genetic analysis.
IMPROVEMENT IN THE ORS OF GENETIC VARIANTS BY DISCOVERY OF SNPS WITH RARE ALLELE FREQUENCY
Much of the speculation about missing heritability from GWASs has focused on the possible contribution of rare variants (low minor allele frequency [MAF] <0.5%). To date, GWAS chips have been created to analyze common variants of >5% MAF. Sequencing of individual genomes has started to generate more information on these rare human chromosome variants.43 The full genome sequences of 1,000 anonymous subjects have been identified. The 1,000 Genomes Project (http://www.1000genomes.org/page.php) has already identified more than 11 million new SNPs in low-depth coverage of 172 individuals. The newly discovered SNPs will be applied to genetic association studies in the near future.
STRUCTURAL VARIATION AND EPIGENESIS IN ASTHMA GENETICS
Previous epidemiological studies demonstrated that SNPs are not responsible for all phenotypical differences. The twin cohort study showed that the concordance rate for self-reported asthma in monozygotic twin pairs is 19%.44 Asthma develops almost concurrently considering that monozygotic twins have similar genetic variants. However, the concordance rate is <20%. One of the explanations for this disconcordance is epigenesis. It was recently discovered that the mother's diet affects the risk of allergic asthma in her offspring.45 Recently, Ho et al.5 extensively reviewed the environmental epigenetics of asthma. Thus, we focused on the epigenesis of aspirin hypersensitivity in asthma. The methylation pattern on the DNA CpG sites was analyzed using a whole genome methylation analysis of 27,168 CpG sites using nasal polyps from patients with aspirin exacerbated respiratory disease (AERD) and aspirin tolerant asthma (ATA).46 As a result, the methylation patterns were significantly different in nasal polyps, but not so different in the buffy coat. A volcano plot showed different methylation levels in AERD and ATA: 332 CpG sites on 296 genes were hypomethylated and 158 sites on 141 genes were hypermethylated (Fig. 2). Hierarchical clustering of 490 differentially methylated CpGs clearly distinguished the nasal polyps between patients with AERD and ATA. The CpG-site methylation of nasal polyps was not correlated with that of the buffy coats, indicating that the difference in methylation pattern was a nasal tissue-specific finding. Among the arachidonate pathway genes, prostaglandin E synthase was hypermethylated and prostaglandin D synthase, arachidonate 5-lipoxygenase activating protein, leukotriene B4 receptor, and lipoxygenase homology domain 1 were hypomethylated (Table 6), indicating that different methylation patterns of these candidate genes in the arachidonate pathway may be responsible for the penetration of specific phenotypes such as AERD in asthma.
Genomic variability is present in many forms, including SNPs, variable number tandem repeats (e.g., mini- and microsatellites), presence/absence of transposable elements (e.g., Alu elements), and structural alterations (e.g., deletions, duplications, and inversions). Until recently, SNPs were thought to be the predominant form of genomic variation and to account for much of the normal phenotypic variation. However, the widespread presence of copy number variation was recently reported in normal individuals.47 Contrary to our previous beliefs, identical twins are not genetically identical. Researchers studied 19 pairs of monozygotic, identical twins and found differences in copy number variations and found that contingent negative variation (CNV) is associated with Parkinson's disease.48 A GWAS of CNV in 16,000 cases of eight common diseases including T1, 2D, rheumatoid arthritis, and Crohn's disease have revealed that some CNV is strongly associated with the risk for complex diseases.
FUTURE OF ASTHMA GENOME RESEARCH
Genome-wide SNP association and fine mapping studies will widen the scope of candidate genes for asthma, but only when applied to sub-phenotypes rather than all asthma types and when analyzed with increased sample sizes considering envienvironmental contributors. An active subject of research in the postgenomic era is to identify functional regulatory elements for human gene expression. Epigenetics is a study of heritable changes in DNA structure that do not alter the underlying sequence. Well-known examples are DNA methylation and histone modification. These changes may remain after cell divisions for the life of the cell and may last for multiple generations. The most exciting idea in epigenetics is that it could be possible to intervene at the junction between the genome and the environment. Epigenetic variants and copy number variations are the starting point to obtain data from clinical samples, and it will be another powerful tool for research into the genetic heritability of asthma.







 XML Download
XML Download