

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Accumulating evidence suggests that obesity-associated inflammation plays an important role in development of obesity-related diseases, such as metabolic syndrome, type 2 diabetes mellitus, and fatty liver disease. The mechanism for the initiation or acceleration of the inflammatory response of adipocytes has not been completely elucidated, however, there is evidence indicating partial involvement of innate pattern recognition receptors in such a process [1,2]. Toll-like receptors (TLRs) are pattern recognition molecules and their signaling pathways are associated with chronic inflammation in obesity [3]. In particular, TLR4 activation leads to up-regulation of interleukin (IL)-6 and release of monocyte chemoattractant protein (MCP)-1 via activation of the extracellular signal-regulated kinase (ERK) [4] and nuclear factor-κB (NF-κB) pathway in adipocytes [5]. In addition, lipopolysaccharide (LPS), a TLR4 ligand, is known as an early key factor that triggers inflammation in adipose tissue [6]. Therefore, LPS-induced TLR4 signaling pathways may be critical factors in adipocyte-related inflammation.
Carnosic acid (CA) is a primary phenolic compound of rosemary (Rosmarinus officinalis) leaves. CA has a wide range of biological activities, including anti-adipogenic, antioxidant, anti-cancer, anti-inflammatory, and neuroprotective effects [7,8,9,10,11]. CA has been shown to decrease body weight gain, fat mass gain, and several inflammatory markers in both diet-induced and genetically modified obesity models [7,8].
CA also effectively attenuates LPS-induced hepatotoxicity via anti-inflammatory and antioxidant activities [9]. CA has also been reported to inhibit adipocyte differentiation by blocking CCAAT/enhancer-binding protein α (C/EBPα) and peroxisome proliferator-activated receptor γ (PPARγ) pathways [10], tumor necrosis factor-α (TNF-α) mediated inflammation and insulin resistance via inhibition of NF-κB, activator protein-1 (AP-1), and forkhead box O1 (FoxO1) signaling in adipocytes [11]. A recent study showed that CA effectively suppresses Gaussia luciferase secretion in 3T3-L1 cells with a secretory Gaussia luciferase gene under control of a NF-κB element [12], however, little is known about the molecular mechanism responsible for its antiinflammatory effect in adipocytes. Therefore, in this study, we investigated the question of whether CA can inhibit LPS-induced TLR4-related signaling in 3T3-L1 adipocytes.
MATERIALS AND METHODS
Materials
CA powder was purchased from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco's modified Eagles medium (DMEM), bovine calf serum (BCS), antibiotic-antimycotic solution, and trypsin-EDTA were purchased from Gibco (Grand Island, NY, USA). LPS, dexamethasone (DEX), and 3-isobutyl-1-methylxanthine (IBMX) were purchased from Sigma-Aldrich (St. Louis, MO, USA). All other reagents were of the highest commercially available grade.
Cell culture and treatment
Mouse 3T3-L1 fibroblasts were obtained from the Korean Cell Line Bank (Seoul, Korea) and cultured in DMEM supplemented with 10% heat-inactivated BCS, 100 U/ml penicillin, and 100 µg/ml of streptomycin. Adipocytic differentiation was induced at 24 h after confluence by adipogenic agents (0.5 mM IBMX, 2 µM DEX, and 1.7 µM insulin) that were added to the culture medium. At day 6-8 of differentiation, 3T3-L1 adipocytes were pre-treated with 5-20 µM CA for 1 h and then stimulated with 100 ng/ml LPS for 30 min. CA was dissolved in dimethyl sulfoxide (DMSO) at a final concentration of 20 µM (or an equivalent volume of DMSO as control) in serum-free medium.
Cell viability (MTT assay)
3T3-L1 cells were seeded at a density of 1 × 105 cells/well in six-well plates. After adipocytic differentiation, mature adipocytes were cultured with various concentrations of CA for 1 h and treated with or without LPS (100 ng/ml) for 30 min. Cells treated with 0.1% DMSO were used as controls. After treatment, the cells were incubated with 5 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution for 4 hours at 37℃. The purple formazan crystals were then dissolved with dimethyl sulfoxide (DMSO). After incubation for 15 minutes, absorbance was measured at 540 nm using a microplate reader (Molecular Devices, CA, USA).
Real-time RT-PCR analysis
Total RNA was isolated from 3T3-L1 adipocytes using Trizol reagent (Invitrogen, CA, USA), following the manufacturer's recommendations. Real-time quantitative PCR was performed using a SYBR Green™ kit (Quantitect™ SYBR Green PCR, Qiagen). The cycling conditions were 15 min at 95℃ and then 40 cycles of 15 s at 94℃ and 30 s at 72℃. Relative quantification was calculated using the ΔΔCT method [13]. Specific primer sets were as follows: 5'-aacatccaaccttcccaaacg-3'/5'-ctcttaacccccgaa tcccag-3' for TNF-α, 5'-tcacctcttcagaacgaattgaca-3'/5'-agtgcctc ttgctgctttcac-3' for IL-6, 5'-attgggatcatcttgctggt-3'/5'-cctgctgtt cacagttgcc-3' for MCP-1, and 5'-gagcgcaagtactctgtgtg-3'/5'-cggactcatcgtactcctg-3' for β-actin.
Western blot analysis
3T3-L1 adipocytes were homogenized in RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% Na-deoxycholate, and 0.1% SDS) containing a protease inhibitor cocktail (Nacalai Tesque) and complete phosphatase inhibitors (Nacalai Tesque). Five micrograms of denatured protein/lane was loaded on polyacrylamide gels, electrophoresed, transferred to polyvinylidene fluoride membranes, and detected using secondary antibodies coupled to horseradish peroxidase. The intensities of the bands were measured using a LAS3000 (Fujifilm, Tokyo, Japan). Antibodies used included anti-ERK (Cell Signaling, Beverly, MA, USA), anti-TLR4 (Santa Cruz Biotechnology, Dallas, TX, USA), and anti-tumor necrosis factor receptor-associated factor 6 (TRAF6) (Santa Cruz Biotechnology, Dallas, TX, USA). β-actin (Cell Signaling, Beverly, MA, USA) was used as the loading control.
Quantification of NF-κB activation
For quantification of NF-κB activity, nuclear extracts were prepared using a Nuclear Extract kit (Active Motif, Carlsbad, CA, USA) and analyzed using a PathScan Total NF-κB p65 Sandwich ELISA kit (Cell Signaling Technology) according to the manufacturer's instructions.
Statistical analysis
Data are expressed as the mean ± standard deviation (SD). All analyses were performed using SAS statistical software version 9.1 (SAS Institute, Cary, NC, USA). Data were analyzed by one-way analysis of variance followed by Duncan's multiple range tests. P < 0.05 was considered statistically significant.
RESULTS
Cell treatment conditions and viability
LPS induced dose-dependent stimulation of IL-6 mRNA expression, which reached significance at a concentration of 100 ng/ml (Fig. 1A). This concentration was chosen for all further incubations in this study for stimulation of IL-6 and ERK and to demonstrate possible inhibitory effects of CA. IL-6 showed high mRNA expression from 30 min of incubation with LPS (Fig. 1B) and ERK was also maximally phosphorylated at 30 min of incubation with LPS (Fig. 1C). Other previous studies have shown that CA has anti-inflammatory effects at 1-30 µM in in vitro study [11,12,14,15]. From these references we selected 10 µM and showed the time-dependent inhibitory effects of CA on IL-6 mRNA expression. As shown in Fig. 1D, incubation with CA (10 µM) effectively inhibited the LPS-induced expression of IL-6 mRNA from 1 hr of incubation. Based on these data we determined the LPS stimulation and the CA treatment conditions.
Cell viability was assessed by MTT assay. As shown Fig. 2, CA in the concentrations used had no effect on viability of 3T3-L1 cells with or without LPS treatment.
Proinflammatory cytokine mRNA levels
Because TNF-α, IL-6, and MCP-1 are major inflammatory cytokines that are overexpressed in adipocytes, we first analyzed the possibility of their induction in response to LPS. As shown in Fig. 3, mRNA expression of pro-inflammatory cytokines was significantly increased by LPS stimulation (P < 0.05). Treatment with CA resulted in a dose-dependent decrease in LPS-induced expression of TNF-α, IL-6, and MCP-1 mRNA compared with that in the LPS control group (Fig. 3A-C, P < 0.05). TNF-α, IL-6, and MCP-1 mRNA expression was significantly decreased by 84.27%, 80.26%, and 72.94%, respectively, in the 20 µM CA group (Fig. 3A-C, P < 0.05).
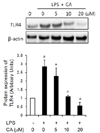
TLR4 protein expression level
For analysis of the effect of CA on TLR4 signals, we analyzed the protein expression level of TLR4. Protein expression of TLR4 was significantly increased by LPS in 3T3-L1 cells, and CA significantly suppressed the protein expression level of TLR4 stimulated with LPS in a dose-dependent manner (Fig. 4, P < 0.05).
MyD88 and TRAF6 protein expression levels
The LPS/TLR4 complex ultimately activates downstream NF-κ B via MyD88- and TRAF6-dependent signaling [16,17]. Results of Western blot analysis showed that LPS treatment caused significantly increased expression of both MyD88 and TRAF6, and this enhancement was suppressed by CA in a dose-dependent manner (Fig. 5A,B, P < 0.05).
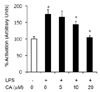
NF-κB activation
Overexpression of MyD88 and TRAF6 leads to activation of NF-κB signaling. Therefore, we assessed NF-κB activation in 3T3-L1 adipocytes and found that NF-κB activation was significantly increased by LPS treatment (175.31%) compared with that in the LPS control group, and this enhancement was significantly decreased in the 10 µM and 20 µM CA groups (Fig. 6, P < 0.05).
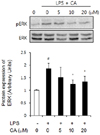
ERK phosphorylation
To gain additional insight into the molecular mechanism underlying the effects of CA, we determined the levels of ERK phosphorylation in induction of proinflammatory cytokine expression. Our results showed that LPS caused upregulated phosphorylation of ERK and CA caused downregulated phosphorylation of ERK in 3T3-L1 adipocytes (Fig. 7).
DISCUSSION
To investigate the potent anti-inflammatory effects of CA in adipocytes we examined the inhibitory effect of CA on the TLR4 signaling pathway in LPS-treated 3T3-L1 adipocytes. First, LPS was applied to differentiated 3T3-L1 adipocytes to induce the TLR4-related inflammatory response. LPS treatment resulted in significantly increased mRNA expression of TNF-α, IL-6, and MCP-1, which was effectively decreased in a dose-dependent manner by treatment with CA. Adipocytes are major producers of inflammatory cytokines and chemokines during development of obesity, and secretion of these proteins leads to insulin resistance and development of metabolic disorders [18,19].
The exact mechanism of the inflammatory response in adipocytes has not been completely elucidated. However, accumulated evidence has shown the partial involvement of innate pattern recognition receptors [1,2]. LPS is an early key factor that triggers inflammation in adipose tissue [6]. Furthermore, a clinical study demonstrated that injection of LPS into healthy volunteers resulted in increased levels of TNF-α and IL-6 in adipose tissue [20]. Other studies have shown that increased endotoxin levels trigger inflammation and body weight gain in an obesity animal model [6]. In addition, during LPS-induced systemic inflammation in mice, adipose tissue was the major source of systemic IL-6 levels [21]. These data suggest that LPS-induced TLR4-related signaling may be a critical factor in obesity-related inflammation. All TLR family members (TLR1-10) are expressed in all human adipose tissue [4]. In particular, TLR4 activation causes upregulation of IL-6 and release of MCP-1 via activation of both the ERK and NF-κB pathways [5]. To examine the mechanism of CA in the inhibition of cytokine gene expression, TLR4 protein expression was measured in LPS-treated 3T3-L1 adipocytes. CA treatment resulted in a dose-dependent decrease in LPS-induced TLR4 protein expression. These data suggest that CA can control TLR4 signaling pathways and these effects may contribute to its inhibitory effect on cytokine gene expression. There are little data on the effect of CA on TLR expression. Recently, an ethanolic rosemary extract containing CA was found to suppress the mRNA expression of TLR2 in Propionibacterium acnes-stimulated monocytic THP-1 cells [22], and TLR4-related indicators were suppressed in RAW264.7 cells [23].
To determine whether the inhibitory effect of CA is mediated by MyD88-dependent pathways, we measured the protein expression levels of MyD88. CA treatment effectively diminished the LPS-induced MyD88 protein up-regulation and caused a significantly decrease in activation of NF-κB and the protein level of TRAF6, which is known to induce phosphorylation of IκB. These data showed that CA can directly suppress MyD88-dependent TLR4 signaling in adipocytes. Furthermore, CA-mediated inhibition of NF-κB activity may affect mitogen-activated protein kinase (MAPK) signaling. The signaling transduction pathway of MAPK plays an important role in transfer of external stimuli into the nucleus through the cytoplasm. The most well known MAPK signaling pathways are ERK, JNK, and p38 [24]. ERK is more strongly phosphorylated than JNK and p38 by LPS treatment in 3T3-L1 adipocytes [25]. Finally, we attempted to determine whether the inhibition of ERK phosphorylation by CA showed an inhibitory pattern similar to that of NF-κB activity. According to the result, treatment with CA resulted in significantly suppressed phosphorylation of ERK, suggesting that the anti-inflammatory action of CA might be mediated by a TLR4-dependent ERK signaling pathway in adipocytes. In adipocytes and adipose tissues, the phosphorylation status of ERK plays a role in modulation of insulin resistance. In obese patients with insulin resistance, pERK and NF-κB, which are active in visceral adipose tissue, are important effectors that promote insulin resistance by controlling inflammatory responses such as the increase of IL-6 secretion in adipose tissues [26]. Recently, Tsai et al. (2014) showed that CA improved TNF-α-induced insulin resistance by decreasing FoxO1, pERK, pJNK, and NF-κB and increasing PPARγ pathway in 3T3-L1 cells [11]. These findings suggested that the anti-inflammatory effect of CA may be attributed to attenuated insulin resistance. In addition, the experimental 1-20 µM carnosic acid levels are equivalent to 0.05 - 1.07 mg/kg/day of carnosic acid. This level is available for intake and is similar to prior used levels in other studies. Previous studies have shown that CA has diverse physiological activities at the dose of 1-30 µM (in vitro) [11,12,13,14] and 1-60 mg/kg (in vivo) [9,27,28]. Therefore, CA may effectively control TLR4-mediated inflammatory disorders in obesity, and the anti-inflammatory effects of CA need to be confirmed in animal models.




 XML Download
XML Download