

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The uncontrolled production of reactive oxygen species (ROS) or reduced cellular antioxidant defense responses may lead to oxidative stress which plays an important role in the oxidation of cellular proteins, lipids, and DNA. Moreover, oxidative stress is responsible for the pathogenesis of aging and degenerative diseases such as cardiovascular disease, neurodegenerative and inflammatory diseases, cancer, and diabetes [1].
Dietary phytochemicals are not only scavengers of free radicals but also modulate pro-survival or pro-apoptotic signaling pathways. It is well known that a number of cellular kinases, such as mitogen-activated protein kinase (MAPK) [extracellular signal-regulated protein kinase (ERK), c-jun N-terminal kinase (JNK) and p38] and phosphoinositide 3 (PI3)-kinase/protein kinase B (Akt) are activated in response to phytochemicals [2]. In previous report, dietary phytochemicals could induce the transcription of phase II antioxidant/detoxifying enzymes through the phosphorylation of MAPKs and PI3K/Akt proteins [3,4]. Therefore, the enhancement of the cellular antioxidant defense potential through the dietary intake of antioxidants is current strategies adopted in the chemoprevention field.
Rice is one of the most commonly consumed food items and a staple in Korean diet. In addition to traditional white rice, a diverse colored rice types such as black, purple, and red rice, which have unique flavor, as well as nutritional and biological properties, have been developed [5]. Previous report has shown that the supplementation of black rice and its anthocyanin pigment can reduce atherosclerotic lesions in hypercholesterolemic animal models [6,7]. Moreover, black rice treatment ameliorated oxidative stress and enhanced antioxidant enzyme activity in HepG2 cells and in C57BL/6 mice [8]. It has also been recently reported that cyanidin-3-O-glycoside (C3G), a typical anthocyanin most abundant in aleurone layer of the black rice, and its metabolites, cyanidin and protocatechuic acid, exert an inhibitory effect on the production of the proinflammatory cytokines in macrophage and mice [9]. Another study has discovered that C3G exerts a protective role against H2O2- or TNFα-induced insulin resistance in 3T3-L1 adipocytes by activating ERK1/2 and PI3K/Akt signaling [10]. As mentioned above, numerous in vitro and in vivo studies have reported on the potential preventive roles against chronic diseases. However, limited studies are available concerning the cytoprotective effects and its possible mechanism of black rice extract against oxidative stress. In this study, we evaluated the protective effect of black rice extract on tert-butyl hydroperoxide (TBHP)-induced oxidative injury in HepG2 cells and also defined several biomarkers that modulate cell survival and death.
MATERIALS AND METHODS
Chemicals
Blakc rice (Oryza sativa cv. Heugjinjubyeo) was purchased from local market (Cheongju, Korea). Extracellular signal-regulated protein kinase 1/2 (ERK1/2) inhibitor (PD98059), Akt inhibitor (LY294002), Caspase-3 assay kit, 2',7'-dichlorofluorescin diacetate (DCFH-DA), tert-butyl hydroperoxide (TBHP), and 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl-terazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Anti-Akt, antiphospho-Akt, anti-ERK1/2, antiphospho-ERK1/2, anti-JNK1/2, and antiphospho-JNK1/2 were obtained from Cell Signaling Technology (Danvers, MA, USA). Fetal bovine serum (FBS), Dulbecco's modified Eagle's medium (DMEM), trypsin-EDTA, and penicillin-streptomycin were obtained from Gibco (Gaithersburg, MD, USA).
Sample preparation
Ground black rice was extracted with methanol by shaking for 24 h at room temperature and filtered through Whatman No. 2 filter paper, and the black rice extract (BRE) was evaporated under vacuum. The dried residue was redissolved in DMSO to a concentration of 100 mg/ml and stored at -20℃ until cellular assay.
Cell culture
HepG2 cells (Korean Collection for Type Cultures) were grown in DMEM containing 10% FBS, 2 mM glutamine, 100 unit/ml penicillin, and 50 µg/ml streptomycin. The cultures were maintained in a humidified incubator with 5% CO2, seeded onto 75-cm2 culture flasks, and harvested with 0.05% trypsin-EDTA.
Assay for TBHP cytotoxicity and protection by BRE
HepG2 cells were seeded in 96-well plates at a density of 1 × 104 cells per well. After 24 h, the culture medium was replaced with FBS-free medium containing various concentration of BRE (10, 50, 100, and 200 µg/ml). After 12 h, the cells were treated for 6 h with 500 µM TBHP to induce oxidative stress. We then evaluated the protective effect of BRE using MTT colorimetric assay [11]. For the MTT assay, the cells were rinsed with PBS twice and MTT (0.5 mg/ml) in FBS-free DMEM was added to the wells. After 4 h incubation, the medium was removed, and blue formazan crystals were dissolved in DMSO and added. We quantified relative cell viability by measuring the absorbance at 550 nm using a microplate reader (BioTek, Inc., Winooski, VT, USA). All experiments were performed in triplicate.
Determination of ROS
Reactive oxygen species (ROS) were quantified with a fluorescent probe DCFH-DA, as previously described [12]. Briefly, cells were seeded in 96-well plates at a density of 5 × 104 cells per well and pretreated with BRE 200 µg/ml. After 12 h, 25 µM DCFH-DA in FBS-free medium was added to the wells for 1 h at 37℃. Then cells were washed with PBS before adding 500 µM TBHP. We measured the fluorescent intensity, corresponding to intracellular ROS, with a fluorescent spectrophotometer (Perkin-Elmer, Norwalk, CT, USA) at an excitation wavelength of 485 nm and an emission wavelength of 530 nm.
Fluorometric analysis of caspase-3 activity
Cells were lysed in a buffer containing 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.4, with 5 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) and 5 mM DTT and assayed for caspase-3 activity according to manufacturer's instructions. The reaction mixture contained 20 mM HEPES (pH 7.4), with 2 mM EDTA, 0.1% CHAPS, 5 mM DTT, and 10 mM Ac-DEVD-AMC (N-acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin) as substrate. Enzymatic activity was measured at an excitation wavelength of 360 nm and an emission wavelength of 460 nm.
Western blot analysis
HepG2 cells were immediately washed with PBS buffer and lysed using a PRO-PREP™ Protein Extraction Solution (iNtRON Biotechnology, South Korea). The protein concentration of each sample was determined using BCA protein assay reagent. Samples were resolved in 8-12% SDS-polyacrylamide gels, followed by electrophoretic transfer to a nitrocellulose membrane. Primary antibodies were applied overnight at 4℃ at a 1:1000 dilution. After incubating with the secondary antibody at room temperature for 2 h, the proteins were detected using the ECL western blotting analysis system (Pierce, Rockford, IL).
Statistical analysis
The results are reported as means ± standard deviation (SD). Significant differences among treatment means were determined using a one-way analysis of variance (ANOVA) followed by Duncan's multiple range test using SAS version 8.1 (SAS Institute, Cary, NC, USA). A p-value of 0.05 was considered significant.
RESULTS
Effect of TBHP on cell viability, caspase-3 activity, and ROS generation in HepG2 cells
The first goal of our study was to determine the oxidative conditions leading to cell death in HepG2. Cells were treated for 0, 3, 6 and 12 h with different concentration of TBHP (50, 100, 200, 500, 1000 µM), and cell viability was determined by MTT assay. Fig. 1A shows that increasing concentrations of TBHP induced a dose-dependent decrease in cell viability. At 6 h, cell death was approximately 60% with a TBHP concentration of 500 µM, where as 1 mM TBHP had lethal effect at 6 h. Therefore, 500 µM TBHP was the concentration chosen for the following experimental conditions.
According to the previous report, TBHP is able to induce the activation of caspase 9 and 3, DNA fragmentation, and eventually, cell death [13]. Therefore, to determine whether the cytotoxic effect of TBHP (500 µM) was due to apoptosis, caspase-3 activity was measured at different times. As shown in Fig. 1B, the 500 µM TBHP induced the activation of caspase-3, which peaked at 6 and 9 h. The results indicate that TBHP activates caspase-3 and induces apoptosis in HepG2 cells.
To elucidate whether TBHP affects the generation of ROS in HepG2 cells and to explain the molecular mechanism of TBHP-induced apoptosis and cell death, we assessed the production of ROS using a fluorescent probe DCFH-DA. As shown in Fig. 1C, 500 µM TBHP that induces apoptosis increased in ROS generation in a time-dependent manner. Our findings suggest that TBHP (500 µM) could induce cell apoptosis and cell death by a rapid increase in intracellular ROS generation.
Protective effect of BRE against TBHP-induced cell death and ROS generation
Before determining the protective effect of BRE, it was necessary to ensure that there was no cytotoxicity. Exposure BRE (10-500 µg/mL) to HepG2 cells over 12 hours incubation period did not decrease cell viability compared to the control and cell viability slightly increased at 200 and 500 µg/mL (Fig. 2A).
To determine the protective effect of BRE against oxidative stress, cells were pretreated with BRE for 12 h and then exposed to 500 µM TBHP for 6 h and then cell death and apoptosis were evaluated. Fig. 2B shows that exposure to 500 µM TBHP for 6 h induced about 70% increase in cell death. However, pretreatment with 10-200 µg/mL BRE for 12 h remarkably decreased cell death in a dose-dependent manner, indicating that BRE-pretreated cells were protected against the oxidative insult. Similarly, Pretreatment of BRE (10-200 µg/mL) prevented the TBHP-induced apoptosis in HepG2 cells at all concentration (Fig. 2C). Since, 200 µg/mL of BRE conveyed a HepG2 with approximately 90% protection against oxidative stress, all subsequent experiments were performed with 200 µg/mL of BRE.
Next, we estimated intracellular ROS production by monitoring changes in DCF fluorescence to evaluate whether BRE attenuates TBHP-induced oxidative stress in HepG2 cells (Fig. 2D). Cells treated with 500 µM TBHP showed a rapid increase in DCF fluorescence intensity compared to non-stressed cells. However, pretreatment with BRE (200 µg/mL) significantly reduced ROS generation.
Effect BRE on the ERKs and JNK activation induced by TBHP.
To elucidate the molecular mechanism involved in HepG2 cell protection by BRE, first, we investigate the effect of TBHP-induced oxidative stress on the activation of ERK1/2 and JNKs. Cells were treated for 1, 3, 6, 9, and 12 h with 500 µM TBHP and protein expression of total and phosphorylated ERKs and JNKs were determined by Western blotting. As shown Fig. 3A, TBHP-induced oxidative stress in HepG2 cells resulted in a transient ERK1/2 activation at 3 and 6 h and a sustained increase of JNK activation.
Next, we investigated the activation of ERKs and JNKs in HepG2 cells pretreated with BRE (200 µg/mL) for 12 h and then exposed to TBHP for different time (0-12 h). In BRE-pretreated cells, ERK1/2 activation peaks at 1h and remained active until 12 h compared to untreated control cell (0 h). However, BRE suppressed TBHP-induced phosphorylation of JNK (Fig. 3B).
Role of ERKs and Akt signaling in the cytoprotective effect of BRE.
To gain further insight into the molecular mechanisms involved in cytoprotective effect of BRE, we attempted to determine whether BRE treatment in HepG2 cells is able to activate key survival and death signals such as ERK1/2, Akt, and JNK in the absence of oxidative stress. As shown Fig. 4, treatments of BRE (200 µg/mL, 12 h) increased the p-Akt and p-ERK protein levels while the levels of p-JNKs were not changed.
Finally, we attempted to determine whether the activation of Akt and ERKs induced by BRE could be involved in protective effect of BRE against TBHP-induced oxidative stress. HepG2 cells were pretreated with 50 µM of PD98059 and LY294002 for 2 h and treated with BRE (200 µg/mL) for 12 h. After then, control and BRE-treated cells were exposed to 500 µM TBHP for 6 and 2 h, respectively, to determine cell viability and intracellular ROS generation. As shown Fig. 5 A and B, pretreatment of BRE significantly reduced the cell death and ROS generation compared to TBHP-treated cells. On the contrary, this effect was partially blunted in the presence of ERKs and Akt inhibitors.
DISCUSSION
This study demonstrates that BRE (black rice extract) has the ability to protect HepG2 cells from oxidative stress-induced cell death by modulating ERK1/2 and Akt activation. We examined the effect of BRE on the response of cultured human HepG2 cells to oxidative stress induced by TBHP. In this study, TBHP, a short-chain analogue of lipid peroxide, was chosen as an oxidative stress inducer because it is well characterized and evokes an increase in intracellular ROS in HepG2 cell lines. Decomposition of TBHP in biological systems will evoke lipid peroxidation chain reactions, induce cell toxicity by damage to DNA, and deplete cell GSH and protein thiols resulting in a general cell damage and apoptosis [14]. The HepG2 cell line was originally derived from a primary hepatoblastoma and has retained the capacity for expressing many plasma proteins that are normally lost in cultured primary hepatocytes. Because of the secretion of many phase I, II, and antioxidant enzymes, cultured HepG2 cell systems are considered a good tool for studying in vitro xenobiotic and cytoprotective mechanisms of natural antioxidants and phytochemicals without using laboratory animals [15]. According to the previous reports, pretreatment of cells with natural phytochemicals prevented the cytotoxicity induced by oxidative stresses [16,17]. Consistent with these reports, our results show that pretreatment of HepG2 cells with BRE attenuated cell death, apoptosis, and intracellular ROS generation induced by TBHP. Although anthocyanin composition of BRE was not determined in this study, previous reports has shown that black rice contained a wide range of total anthocyanin content, with cyanidin 3-glucoside being the most common anthocyanin, whereas peonidin-3-glycoside was the second dominant anthocyanin [18]. Therefore, we hypothesized that anthocyanins such as cyanidin-3-glycoside and peonidin-3-glycoside contained in the black rice extract might be the major component responsible for the protective effects in the present study. However, it cannot be ruled out that flavonoids or phenolic acid in the extract may contribute partially to protective effects observed in this study.
Oxidative stress-induced injury results not only from direct chemical interactions by oxidizing cellular proteins, lipids, and DNA, but also from alterations in signal transduction pathways. To elucidate the molecular mechanism involved in HepG2 cell protection by BRE, we investigate the effect BRE pretreatment on the activation of ERK1/2 and JNKs which are key mediators of stress signals and might be responsible for protective mechanism and stress-induced apoptosis [19]. ERKs are major signaling enzymes involved in cell survival against oxidative stress. Unlike the ERK pathway, the JNK cascade is activated by stress signals such as UV, γ-irradiation, free radicals, and inflammatory cytokines Therefore, JNK is also known as stress-activated protein kinase [4]. As shown Fig. 3B, in BRE-treated cells before induction of oxidative stress, ERK1/2 activation peaks at 1h and remained active until 12 h compared to BRE-untreated cells. However, BRE suppressed TBHP-induced phosphorylation of JNK. It is thought that the protective effect of BRE on TBHP-induced cell death might be mediated through the sustained activation of ERKs and the prevention of prolonged JNK activation.
In previous reports, dietary antioxidant can activate a number of cellular kinases, including MAPKs and PI3K/Akt which are responsible for cell survival against oxidative stress [20]. As shown Fig. 4, treatments of BRE increased the p-Akt and p-ERK protein levels while the levels of p-JNKs were not changed. Previous studies reported that cyanidin 3-glucoside, a typical anthocyanin most abundant in black rice, exerts a protective effect against H2O2- or TNF-α-induced insulin resistance in adipocytes by inhibiting the JNK signaling pathway [10]. Moreover, bayberry extracts containing 95% cyanidin 3-glucoside protect β cells from H2O2-induced cell injury by activating ERK1/2 and PI3K/Akt signaling [21].
Finally, to investigate the signaling pathway involved in the protective effect of BRE, we explored the Akt and ERK pathways using the appropriate inhibitors, LY294002 and PD98059, respectively. The results showed that ERKs and Akt inhibitors blunted the protective effect of BRE on TBHP-induced cell death and ROS generation. Taken together, our results suggest that activation of ERKs and Akt pathway might be involved in the cytoprotective effect of BRE against oxidative stress.
In conclusion, our results show that pretreatment of HepG2 cells with BRE strongly protects the cells against TBHP-induced oxidative stress by reducing cell death, caspase-3 activity, and ROS generation and also by preventing ERK1/2 deactivation and the prolonged JNK activation. Moreover, pretreatment of BRE increased the activation of ERKs and Akt which are pro-survival signal proteins. Based on the results from ERKs and Akt inhibitor experiments, we hypothesized that that activation of ERKs and Akt pathway might be involved in the cytoprotective effect of BRE. While, as discussed above in regard to the complexity of composition of black rice extracts, further studies are needed to clearly elucidate which particular compounds are responsible for the protective effect. Our findings provide new insights into the cytoprotective effects and its possible mechanism of black rice against oxidative stress.




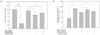
 XML Download
XML Download